Neurodegenerative Erkrankungen
Neurodegenerative Erkrankungen
Verlauf
Bei guter Verträglichkeit wurde Pramipexol bei Progredienz der Symptome über einen Zeitraum von drei Jahren auf 3,15 mg/d erhöht und zudem um den MAO-Hemmer Rasagilin 1 mg/d ergänzt, bei weiter guter Verträglichkeit. Ein Jahr nach der letzten Dosiserhöhung berichtete die Ehefrau auf Nachfrage, dass der Patient vermehrt esse und am PC stundenlang erotische Webseiten besuche. Unter der Annahme einer Impulskontrollstörung wurde Pramipexol sofort abgesetzt und auf eine Monotherapie mit Levodopa (bis 1.000 mg/d) umgestellt. Darunter war die Symptomatik rasch regredient, allerdings klagte der Patient nun über Affektlabilität und depressive Stimmung. Zudem traten nun trotz fünf bis sechs Einnahmezeitpunkte von Levodopa vermehrt Fluktuationen der Beweglichkeit, insbesondere mit starken Off-Phasen auf. Es erfolgte eine zusätzliche antidepressive Behandlung mit Venlafaxin (150 mg/d). Nach vier Monaten wurde niedrigdosiert wieder ein Dopaminagonist (Rotigotin t.d. 4 mg/d) ergänzt, außerdem wurde mit dem Patienten die Therapieoption einer „Tiefen Hirnstimulation“ (THS) erstmals besprochen.
Beschreibung
Dieser Fall beschreibt die hochrelevante Nebenwirkung einer Impulskontrollstörung bei einem IPS unter Dopaminagonisten-Behandlung. In der Initialtherapie des IPS werden MAO-B-Hemmer, Dopaminagonisten oder Levodopa eingesetzt [1]. Bei jüngeren Patienten (< 70 Jahre) werden dabei neben einem MAO-B-Hemmer bevorzugt die nicht-ergolinen Dopaminagonisten Pramipexol, Ropinirol, Rotigotin oder Pirepidil verwendet. Da Dopaminagonisten auch an periphere Dopaminrezeptoren binden, können darunter periphere Nebenwirkungen wie Knöchelödeme, orthostatische Beschwerden und Übelkeit auftreten. Außerdem besteht ein erhöhtes Risiko für neuropsychiatrische Symptome wie Impulskontrollstörungen, Halluzinationen oder ein dopaminerges Dysregulationssyndrom. Impulskontrollstörungen sind in der klinischen Praxis immer noch unterdiagnostiziert, deren Erkennen jedoch hochrelevant. Zu den typischen Impulskontrollstörungen bei IPS-Patienten gehört neben pathologischem Spielen, exzessives Einkaufen, maßloses Essen und Hypersexualität. In bis zu sechs Prozent treten sie kombiniert auf. Esssucht und Hypersexualität kommen dabei doppelt so häufig vor [3]. Auch Verhaltensauffälligkeiten mit exzessiv wiederholten Tätigkeiten und zwanghafter Medikamentengebrauch sollten regelmäßig erfragt werden. [2, 3]. Die Pathophysiologie ist bisher ungeklärt, es scheint ein komplexer Mechanismus mit Beteiligung des dopaminergen, serotonergen und noradrenergen Systems mit Imbalance des Belohnungs- und Verhaltenssystems vorzuliegen. Insbesondere kann aus der höheren Dopamin-D3-Rezeptor-Affinität von Dopaminagonisten im mesolimbischen System eine Überstimulation des Dopamin-D3-vermittelten kurzfristigen Belohnungslernen resultieren [2, 3, 5].
Die Risikofaktoren für die Entwicklung einer Impulskontrollstörung sind in Tabelle 1 zusammengefasst. Das Auftreten von Impulskontrollstörungen ist abhängig von Dauer und Dosis der Dopaminagonistentherapie, aber auch von Geschlecht und Alter der Patienten [3]. Es wird diskutiert, ob die Grunderkrankung an sich mit einem erhöhten Risiko für Impulskontrollstörungen assoziiert ist. Die Prävalenz bei behandelten Patienten steigt auf bis zu 30 Prozent nach fünf Jahren an. Wie im aktuellen Fall können sie erst mehrere Jahre nach Therapiebeginn auftreten [3, 4]. Der erste Schritt besteht daher immer aus Prävention mit sorgfältiger Aufklärung von Patienten und Angehörigen sowie regelmäßiger Nachfrage der Symptome auch nach Jahren guter Verträglichkeit. Durch Reduktion oder Beendigung der Therapie sind die Symptome meist gut rückläufig [3]. Komplizierend kann ein Dopaminagonisten-Entzugssyndrom mit depressiver Störung, Panikattacken, Reizbarkeit und autonomen Symptomen auftreten. Eine Umstellung auf einen anderen Dopaminagonisten kann nach einigen Monaten Symptomfreiheit unter engmaschiger Kontrolle versucht werden. Unterstützend kann eine niedrigdosierte Behandlung mit Clozapin oder Quetiapin begonnen werden, gleiches gilt für eine Psychotherapie. Der Einsatz der frühen THS besonders im Nucleus subthalamicus wird unter anderem wegen der dabei meist möglichen Reduktion der dopaminergen Gesamtdosis diskutiert [5, 6].

Tabelle 1: Risikofaktoren für Impulskontrollstörungen beim idiopathischen Parkinson-Syndrom (modifiziert nach [1 und 2]).
Zusammenfassend können Impulskontrollstörungen bei IPS-Patienten zu schwerwiegenden finanziellen, psychosozialen oder rechtlichen Konsequenzen führen. Daher sind Patientenaufklärung und regelmäßige klinische Kontrollen wichtig, da es sich hierbei um eine einfach zu diagnostizierende und behandelnde Nebenwirkung handelt.
Akutes Delir bei Demenz vom Alzheimer-Typ
Anamnese
Ein 72-jähriger Mann wurde vom Rettungsdienst wegen eines akuten Verwirrtheitszustands in die neurologische Notfallaufnahme gebracht. Im häuslichen Umfeld hatte er sich in den Tagen zuvor zunehmend desorientiert gezeigt und den nicht im Haushalt lebenden Sohn teilweise nicht erkannt. Zudem zeigte er wiederholt illusionäre Verkennungen, bei denen er Bäume im Garten für fremde Personen hielt. Die Ehefrau berichtete, dass der Patient seit dem vergangenen Jahr zunehmende Gedächtnisstörungen entwickelt hatte, insbesondere konnte er sich wiederholt an Vereinbarungen und Termine nicht erinnern und verlief sich zweimal auf dem Rückweg von der nahen Sporthalle. Neben einem arteriellen Hypertonus, der mit Ramipril behandelt war, bestanden keine weiteren Vorerkrankungen.
Diagnostik
Bei Aufnahme war der Patient nur zur Person orientiert, wirkte unruhig, ratlos und teilweise misstrauisch. Zudem deutete er wiederholt auf eine Ecke des Raums, in der sich aber keine Person befand. Ansonsten zeigte er in der neurologischen Untersuchung kein fokal-neurologisches Defizit. Die Schleimhäute waren trocken. In der Notfallbildgebung (CCT, Abbildung 2) konnte eine Akutpathologie ausgeschlossen werden, jedoch zeigte sich eine erheblich generalisierte Hirnatrophie mit deutlicher temporaler Betonung. Die Labordiagnostik ergab eine Infektkonstellation (Leukozytose mit 11.300/µl, C-reaktives Protein 56 g/dl) bei einem Nitrit-positiven Harnwegsinfekt.
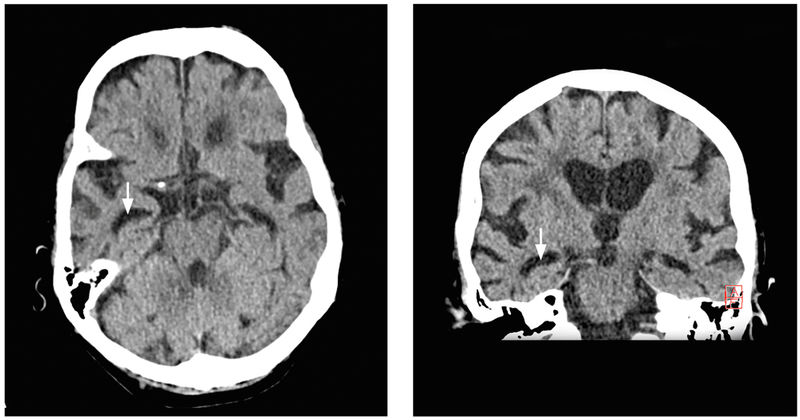
Abbildung 2: Temporal betonte Hirnatrophie im kraniellen CT (axial links, koronare Rekonstruktion rechts). Mit freundlicher Genehmigung von Professor Dr. C. Wendl, Universitätsklinikum Regensburg
Verlauf
Unter der Diagnose einer infektassoziierten Dekompensation einer bestehenden Demenz erfolgte die stationäre Aufnahme zur antibiotischen Therapie mit Amoxicillin/Clavulansäure und zur Volumentherapie. Da der Patient in der Akutphase nicht einwilligungsfähig war, wurde die Einrichtung einer gesetzlichen Betreuung für die Bereiche Gesundheitsfürsorge und Aufenthaltsbestimmung mit der Ehefrau als vorläufige Betreuerin angeregt und bewilligt.
Parallel zur antibiotischen Behandlung wurde eine niedrigdosierte antipsychotische Behandlung mit Risperidon 0,5 mg zweimal täglich begonnen. Innerhalb weniger Tage besserte sich die delirante Symptomatik, sodass Risperidon wieder abgesetzt werden konnte. Mit dem nun wieder einwilligungsfähigen Patienten wurde, unterstützt durch die Ehefrau, eine erweiterte Diagnostik hinsichtlich seiner demenziellen Entwicklung vereinbart. Dabei erfolgte eine ausführliche neuropsychologische Testung, die vor allem eine Einschränkung von Gedächtnis und räumlicher Verarbeitung/Orientierung sowie eine Werkzeugstörung zeigte, insgesamt passend zu einer Demenz vom Alzheimer-Typ. Die zudem erfolgte Liquordiagnostik ergab (bei unauffälligen Akutparametern) leicht erhöhte Tau-Werte sowie eine beta-Amyloid-Ratio von 0,85 (Norm < 0,5). Zur Stabilisierung wurde Rivastigmin, zunächst mit 4,6 mg/Tag eindosiert. Außerdem erfolgte eine ausführliche Beratung des Patienten und der Ehefrau bezüglich nicht-medikamentöser Verfahren und Hinweisen für die weitere Betreuung des Patienten, der schließlich nach sieben Tagen wieder ins häusliche Umfeld entlassen werden konnte.
Beschreibung
Die Demenz vom Alzheimer-Typ ist die häufigste neurodegenerative Erkrankung mit einer Prävalenz in Deutschland von 1,9 Prozent [7]. In der Gruppe der über 80-jährigen sind über 20 Prozent von einer Demenz vom Alzheimer-Typ betroffen. Grundsätzlich nimmt das Risiko, an einer Alzheimer-Demenz zu erkranken mit steigendem Alter zu. Früh beginnenden Demenzen (< 60 Jahre) liegt gelegentlich eine erbliche Ursache (unter anderem Mutationen im Presenilin oder Amyloid-Precursor-Protein-Gen) zugrunde. Als Instrumente zur orientierenden Einschätzung von kognitiven Störungen sind zum Beispiel der Mini-Mental-Status-Test (MMST), DemTect und der Montreal-Cognitive-Assessment-Test (MoCA) geeignet. Der Uhrentest (Abbildung 3), der insbesondere visuokonstruktive Funktionen testet, kann in Kombination mit den anderen genannten Kurztestverfahren die diagnostische Aussagekraft erhöhen. Die Diagnose basiert auf einer sorgfältigen Anamneseerhebung sowie der gezielten neuropsychologischen Testung, zudem kann die bildgebende Diagnostik (MRT, CT) relevante Differenzialdiagnosen ausräumen. Die zusätzliche Liquordiagnostik stützt die Diagnose [8]. In unklaren Fällen kann eine zerebrale FDG-PET die Diagnose sichern [9].
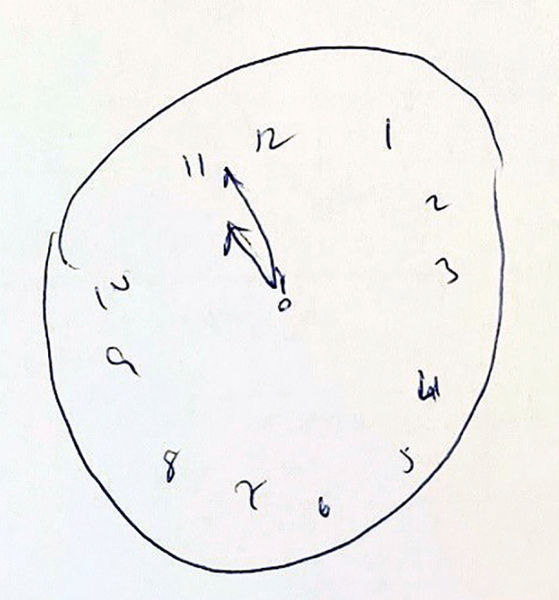
Abbildung 3: Uhrentest nach Shulman mit fehlerhaft eingezeichnetem Zeiger.
Zur Unterstützung der kognitiven Funktion ist eine Behandlung mit AcetylcholinesteraseInhibitoren Rivastigmin, Donezepil und Galantamin etabliert. Zur Behandlung bei Patienten mit mittelschwerer bis schwerer Demenz kann auch Memantine eingesetzt werden. Die nicht-medikamentöse Therapie beinhaltet unter anderem Gedächtnistraining, Ergo-, Logo- und Physiotherapie. Die Entwicklung neuer Therapien, die eine Verlangsamung der progredienten Neurodegeneration bei der Alzheimer-Demenz zum Ziel haben, verlief in den vergangenen zehn Jahren enttäuschend. Umso größere Aufmerksamkeit erhielt in diesem Jahr eine vorläufige Zulassung des gegen aggregiertes Amyloid-beta gerichteten Antikörpers Aducanumab in den USA durch die FDA (Food and Drug Administration; [10]) auf der Grundlage von Surrogat-Endpunkten einer Reduktion der Amyloid-Plaques. Eine europäische Zulassung steht bislang aus.
Exsikkose, bakterielle und virale Infekte oder Verschlechterung der Nierenfunktion begünstigen also eine Exazerbation der Symptome einer Demenz bis hin zum Delir. Deshalb ist, wie im vorliegenden Fall, eine rechtzeitige und konsequente Behandlung von Infekten notwendig.
Progrediente Fußheberschwäche
Anamnese
Eine 53-jährige Frau stellte sich mit einer seit einem Jahr progredienten Fußheberschwäche rechts vor. Aufgrund dessen hatte sie eine operative Mikrodekompression LWK 4/5 erhalten, allerdings ohne Besserung. Es zeigten sich in der klinischen Untersuchung eine mittelgradige distal betonte Beinparese mit Muskelatrophien des rechten Beins, jedoch auch eine leichte Fußheberschwäche links und lebhafte Muskeleigenreflexe mit verbreiterten Reflexzonen, ohne Pyramidenbahnzeichen.
Diagnostik
Elektromyografisch fand sich chronisch-neurogener Umbau in der Muskulatur des rechten Arms und beider Beine sowie paravertebral, zusätzlich konnten in drei verschiedenen Regionen akute Denervierungszeichen detektiert werden (Abbildung 4). Ergänzend ergaben sich in der transkraniellen Magnetstimulation Hinweise für eine Schädigung der Pyramidenbahn zu beiden Beinen. Darüberhinausgehende Pathologien (Polyneuropathie oder Leitungsblöcke) fanden sich nicht. Eine spinale und zerebrale MRT zeigten keine erklärenden Ursachen.
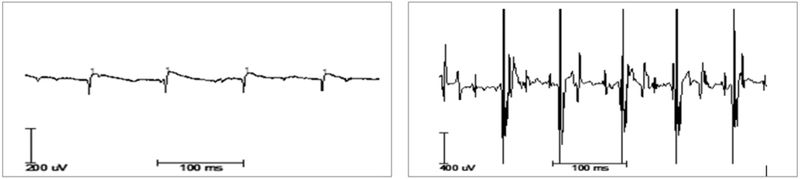
Abbildung 4: Nadel-EMG-Untersuchung mit Zeichen einer floriden neurogenen Schädigung mit positiv scharfen Wellen (spontan, links) und chronisch neurogenen Umbaus mit erhöhten Potenzialen (Willkür, rechts).
Verlauf
Unter der Diagnose einer Motoneuronerkrankung im Sinne einer Amyotrophen Lateralsklerose (ALS) wurde eine Therapie mit Riluzol initiiert. Dennoch entwickelte die Patientin innerhalb eines Jahres eine hochgradige beinbetonte Tetraparese mit Dysarthrie.
Beschreibung
Die Diagnose der ALS (Inzidenz etwa 3/100.000) erfolgt durch Anamnese und klinisch-neurologischen Befund, unterstützt durch neurophysiologische Untersuchungen. Unspezifische Erstsymptome, insbesondere bei jüngeren Patienten ≤ 45 Jahre, können die rechtzeitige Diagnose jedoch erschweren, wohingegen eine bulbäre Symptomatik mit einer früheren Diagnosestellung assoziiert ist [11]. Die protrahierte Diagnosestellung der ALS kann im Schnitt zwölf Monate und somit bereits ein Drittel der Erkrankungszeit (Überlebensdauer drei bis fünf Jahre) betragen [12].
Klassischerweise liegt eine Schädigung beider Motoneurone vor (Tabelle 2), Varianten mit (inital) vorwiegendem Betroffensein nur eines Motoneuronsystems sind jedoch möglich. Kennzeichnend ist die rasche Progression der motorischen Symptomatik, die letztlich zur zunehmenden Gangunfähigkeit, Kommunikationsverlust durch Dysarthrophonie, Dyspnoe sowie Gewichtsverlust durch Dysphagie und Atrophie führt. Darüber hinaus können auch nicht-motorische Symptome wie eine fronto-temporale Demenz (ALS-FTD) und pathologisches Lachen oder Weinen vorliegen. Die Differenzialdiagnostik sollte sorgfältig erfolgen, um behandelbare Ursachen wie zerebrale Raumforderungen, entzündliche Erkrankungen, Ischämien, Spinalkanalstenosen mit Myelopathie bzw. Neuropathien auszuschließen.
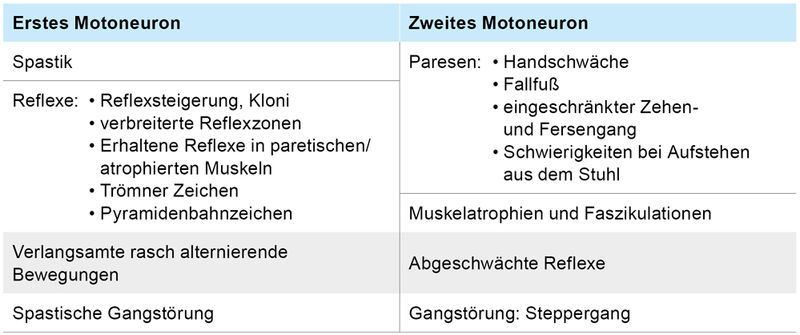
Tabelle 2: Klinische Zeichen einer Schädigung des ersten und zweiten Motoneurons bei Motoneuronerkrankungen.
Aufgrund fehlender kurativer Therapien stehen aktuell krankheitsmodifizierende Behandlung, Symptomlinderung (Tabelle 3) und eine den klinischen Einschränkungen entsprechende begleitende Physiotherapie, Ergotherapie and Logopädie im Vordergrund [15, 16]. Eine adäquate Hilfsmittelversorgung zum größtmöglichen Erhalt der Autonomie sollte frühzeitig eingeleitet werden. Zugelassen ist der Glutamatantagonist Riluzol, welcher zu einer signifikanten Lebensverlängerung zwischen sechs bis 19 Monaten führt [14]. Weitere Medikamente zeigten in klinischen Studien bisher leider keine signifikante Effektivität.
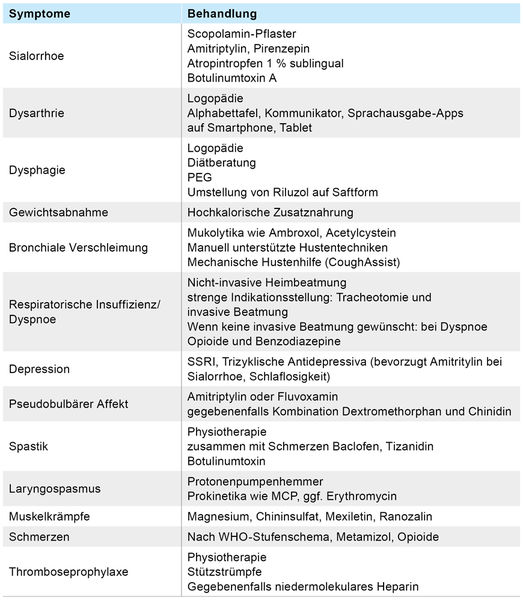
Tabelle 3: Übersicht der symptomatischen Therapie bei ALS-Patienten (nach Leitlinie DGN 2021).
Am meistens profitieren Patienten bezüglich Lebenserwartung und -qualität von einer Behandlung in einer ALS-spezialisierten Klinik mit einem multidisziplinären Team aus Neurologie, Palliativmedizin, Psychologen und Therapeuten. Es soll betont werden, dass im Endstadium der ALS insbesondere die Dyspnoe eine hohe Prävalenz aufweist. Bei ersten Hinweisen auf eine respiratorische Insuffizienz ist eine Vorstellung in einer Lungenfachklinik indiziert, um frühzeitig eine nicht-invasive Heimbeatmung in die Wege leiten zu können, welche mit einem verlängerten Überleben assoziiert ist [17]. Im späteren Verlauf ist eine palliativmedizinische Betreuung notwendig, hier muss rechtzeitig eine dem Patientenwunsch entsprechende Versorgung eingeleitet werden. Dementsprechend erfolgen frühzeitig vor dem Auftreten von Kommunikationsproblemen die Patientenaufklärung und das Erstellen einer Patientenverfügung.
Das Literaturverzeichnis kann im Internet unter www.bayerisches-aerzteblatt.de (Aktuelles Heft) abgerufen werden.
Die Autoren erklären, dass sie keine finanziellen oder persönlichen Beziehungen zu Dritten haben, deren Interessen vom Manuskript positiv oder negativ betroffen sein könnten.
Das Wichtigste in Kürze
Bei jüngeren Patienten mit IPS werden MAO-B-Hemmer und bevorzugt nicht-ergoline Dopaminagonisten verwendet.
Impulskontrollstörungen als Nebenwirkung einer Dopaminagoinisten-Therapie sind in der Regel gut beherrschbar, müssen aber regelmäßig erfragt werden, um schwerwiegende Konsequenzen zu vermeiden.
Demenzen werden anhand der klinischen Symptomatik ätiologisch eingeordnet. Die weitere Diagnostik basiert auf neuropsychologischer Testung und Bildgebung (MRT, CT). Die zusätzliche Liquordiagnostik und zerebrale FDG-PET können vor allem die Diagnose einer Demenz vom Alzheimer-Typ stützen.
An erster Stelle der Akuttherapie bei einem demenzassoziierten Delir steht die Ursachenbehandlung (Infekt, Exsikkose) sowie Maßnahmen zur Reorientierung und Angstreduktion.
Bei progredienten, asymmetrischen Paresen mit Muskelatrophie sollte an eine Motoneuronerkrankung gedacht und früh an eine/n Neurologin/Neurologen überwiesen werden.
Die Therapie der amyotrophen Lateralsklerose stützt sich auf eine progressionsverzögernde Therapie mit Riluzol sowie auf symptomatische Therapien, vor allem Physio- und Ergotherapie sowie Logopädie und eine rechtzeitige Hilfsmittelversorgung. Im späteren Verlauf ist eine palliativmedizinische Betreuung notwendig, hier muss rechtzeitig eine dem Patientenwunsch entsprechende Versorgung eingeleitet werden.
Autoren
Dr. Ohnmar Hsam
Privatdozent Dr. Zacharias Kohl
Klinik und Poliklinik für Neurologie der Universität Regensburg,Am medbo Bezirksklinikum Regensburg, Universitätsstr. 84, 93053 Regensburg