05.05.2021
Titelthema
![]() Artikel als PDF
Artikel als PDF
Kinderkardiologie – highlighted
 Kinderkardiologie – highlighted
Kinderkardiologie – highlighted
In Deutschland leben weniger als ein Prozent der Menschen mit einem angeborenen Herzfehler. Die Behandlungserfolge in der Kinderkardiologie in den vergangenen Jahrzehnten haben mittlerweile aber auch in der Erwachsenenmedizin eine besondere Patientengruppe entstehen lassen: Erwachsene mit einem angeborenen Herzfehler (EmaH). In der Neonatologie geht es vor allem um Notfallversorgung und die rasche Diagnose angeborener Herzfehlbildungen, in anderen Disziplinen der Pädiatrie sind angeborene Herz- und Gefäßfehlbildungen und Herzkreislauferkrankungen wichtige Differenzialdiagnosen verschiedener respiratorischer und infektiologischer Krankheitsbilder und Symptome. Die enge Kooperation der Kinderkardiologie mit der spezialisierten Herzchirurgie angeborener Herzfehler ist historisch gewachsen und gefestigt. Operative und interventionelle Methoden sind in der Behandlung komplexer angeborener Herzfehler eng miteinander verbunden.
Fall 1: Häuslicher Notfall bei einem reifen Neugeborenen mit kritischer Aortenisthmusstenose
Anamnese
Tim ist das erste Kind einer 33-jährigen Erstgravida. Nach regelrecht verlaufener Schwangerschaft unter Wahrnehmung aller Vorsorgetermine kommt Tim zwei Stunden nach spontanem Blasensprung in der 41. Schwangerschaftswoche zur Welt. Die Entbindung erfolgt in einer Geburtsklinik; Geburtsgewicht 3,1 kg, APGAR-Werte 7/9/10. Mutter und Kind sind wohlauf und können die Geburtsklinik bereits nach vier Stunden verlassen. Der erste Lebenstag zuhause verläuft harmonisch. Tim trinkt erste Schlucke an der Brust und setzt Urin und Mekonium ab. Am zweiten Lebenstag findet die Hebamme bei einem Hausbesuch das Kind blass, zyanotisch mit „knorzender“ Atmung vor. Sie erkennt die Notfallsituation und ruft unverzüglich den Babynotarzt.
Diagnostik und Therapie
Der Notarzt bringt das Kind, mit einer peripheren Infusion und Glucose 5%-Infusion versorgt, unverzüglich in die nächstgelegene Kinderklinik. Dort wird Tim intubiert und beatmet. Unter dem primären Verdacht auf eine Neugeborenensepsis wird eine antibiotische Therapie begonnen. Das Thorax-Röntgenbild zeigt eine pulmonale Stauung. Ein systolisches Herzgeräusch, niedrige Blutdruckwerte und schwache Leistenpulse bei einer gemischt respiratorischen und metabolischen Azidose führen zur Verdachtsdiagnose eines Vitums cordis congenitum.
Die Echokardiografie bestätigt den Verdacht und zeigt eine kritische Aortenisthmusstenose mit schlechter linksventrikulärer Funktion, Mitralklappeninsuffizienz und Lungenstauung (Abbildung 1A und 1B). Um den Ductus arteriosus wiederzueröffnen, erhält Tim nun eine hochdosierte Prostaglandin E2-Infusion und wird in das Kinderherzzentrum weiterverlegt. Bei unter Prostaglandin E2-Therapie wiedereröffnetem Ductus wird Tim mit Katecholaminen behandelt und nach partieller Erholung der linksventrikulären Funktion zwei Tage später mit einer extended resection und End-zu-End-Anastomose der Aorta descendens mit dem Aortenbogen operiert. Er benötigt noch zwei Tage Erholung, bevor er vom Respirator entwöhnt werden kann. Nach zögerlichem Nahrungsaufbau kann er 14 Tage später die Klinik verlassen.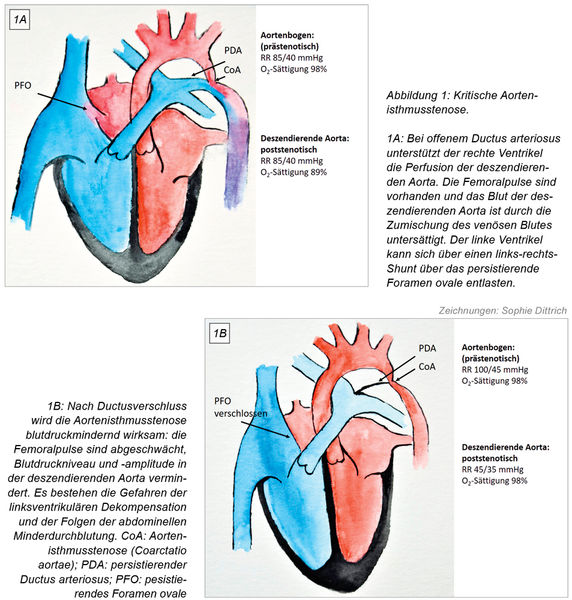
Diskussion
Bei Tim hätte eine intrauterine Diagnose oder die Durchführung des Pulsoxymetrie-Screenings die häusliche Dekompensation bei Ductusarteriosus-Verschluss wahrscheinlich verhindert [1]. Bei intrauterin bekannter Diagnose wäre unmittelbar nach der spontanen Entbindung mit einer niedrigdosierten Prostaglandin E2-Medikation zum Offenhalten des Ductus begonnen worden [2, 3]. Die Operation hätte elektiv stattfinden, die präoperative Dekompensation und die lange postoperative Erholung hätten mit großer Wahrscheinlichkeit vermieden werden können.
Ductusabhängige Herzfehler bei Neugeborenen
Der fetale Kreislauf weist in der Regel über das Foramen ovale und den Ductus arteriosus Shunt-Verbindungen auf Vorhof- und Gefäßebene auf, die dem Herzen im fetalen Kreislauf die Möglichkeit geben, eine Vielzahl komplexer Herz- und Gefäßfehlbildungen zu kompensieren [3, 4]. Sowohl bei einer Ductus arteriosus-abhängigen Lungenperfusion wie zum Beispiel bei Trikuspidal- oder Pulmonalatresien, als auch bei Ductus arteriosus-abhängigen Systemperfusionen wie zum Beispiel der kritischen Aortenisthmusstenose oder dem hypoplastischen Linksherzsyndrom mit Mitral- und Aortenatresie, ist das fetale Gedeihen oft unbeeinträchtigt. Meistens persistiert unmittelbar nach der Entbindung ein Shunt über das Foramen ovale und den Ductus arteriosus. Klinische Symptome des angeborenen Herzfehlers können dann unter Umständen sehr milde ausgeprägt sein oder gar ganz fehlen. Die klinische Symptomatik entwickelt sich dann erst mit dem physiologischen Abfall des Lungengefäßwiderstandes (-> Zunahme des Shuntvolumens in den Lungenkreislauf) und/oder dem Verschluss des Ductus arteriosus (-> zunehmende, unter Umständen kritische Einschränkung der Aorten- oder Pulmonalarteriendurchblutung).
Pulsoxymetrie-Screening
Seit 2017 ist das Pulsoxymetrie-Screening in das Vorsorgeprogramm des Neugeborenen eingeführt. Die pulsoxymetrische Sauerstoffsättigungsmessung am Fuß des Neugeborenen soll möglichst 24 bis 48 Stunden nach Geburt, frühestens vier Stunden danach vorgenommen werden. Für Hausgeburten wird die Pulsoxymetrie-Untersuchung spätestens bei der U2, also zwischen dem dritten und zehnten Lebenstag empfohlen. Die U2 ist die empfohlene erste ärztliche eingehende körperliche Grunduntersuchung, bei der angeborene Erkrankungen und Fehlbildungen (zum Beispiel Fehlbildungen des Herzens) erkannt und lebensbedrohliche Komplikationen vermieden werden sollen. Beim Pulsoxymetrie-Screening gilt eine mindestens 96-prozentige Sauerstoffsättigung als unauffälliges Ergebnis, Werte zwischen 90 und 96 Prozent sollen innerhalb von zwei Stunden wiederholt werden. Liegt bei einer wiederholten Messung der Messwert weiter unter 96 Prozent oder liegt der erste Messwert unter 90 Prozent, so ist unverzüglich eine weitere Diagnostik und nach Ausschluss sonstiger Ursachen eine unverzügliche Herzultraschalldiagnostik indiziert [1].
Fall 2: Hypoxämischer Anfall bei einem Säugling mit Fallot-Tetralogie
Anamnese
Die Diagnose Fallot-Tetralogie wurde bei Fabian schon intrauterin gestellt. Die Entbindung erfolgte in der 39. Schwangerschaftswoche nach spontaner Geburt; Geburtsgewicht 3.150 g, APGAR 8/9/9. Die Echokardiografie bestätigte die Diagnose der Fallot-Tetralogie mit den anatomischen Details eines elongierten, mit 5 mm Durchmesser hypoplastischen, rechtsventrikulären Ausflusstraktes, einer bikuspiden, mit 6 mm ebenfalls hypoplastischen und stenotischen Pulmonalklappe und einer mäßigen Hypoplasie der zentralen Pulmonalgefäße. Initial bestand ein noch offener Ductus arteriosus, der bei einer echokardiografischen Kontrolle am sechsten Lebenstag aber verschlossen war. Am Ende der ersten Lebenswoche lag die Sauerstoffsättigung des Kindes in der stationären Überwachung bei Werten zwischen und 80 und 92 Prozent. Fabian wurde vollgestillt und hatte bereits am Ende der ersten Lebenswoche sein Geburtsgewicht wieder erreicht. Bei echokardiografisch messbarer Zunahme der Blutflussgeschwindigkeit im rechtsventrikulären Ausflusstrakt begann der betreuende Kinderkardiologie ambulant eine Therapie mit einem Beta-Blocker [5]. Bei einer elektiven ambulanten Vorstellung in der kinderkardiologischen Praxis im Alter von sechs Wochen kam es in der Untersuchungssituation zu einem Sauerstoffsättigungsabfall mit sichtbarem, blass bläulich fahlem Hautkolorit. Mit der Diagnose hypoxämischer Anfall veranlasste der Kinderkardiologe die unverzügliche Einweisung in das Herzzentrum. 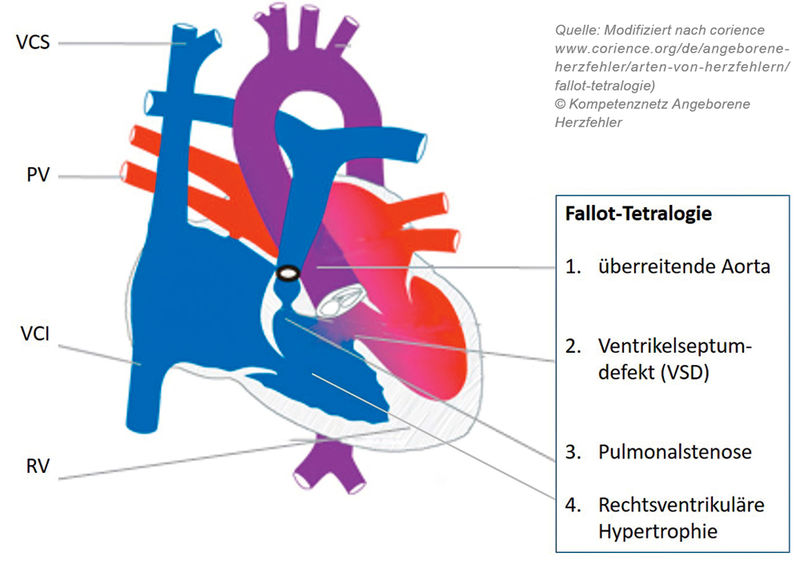
Diagnostik und Therapie
Auf der Station zeigte Fabian in der pulsoxymetrischen Überwachung Sauerstoffsättigungswerte um 85 bis 88 Prozent, anfallsweise aber auch Entsättigungsperioden bis 65 Prozent, sodass er eine Sauerstoffvorlage (1 Liter/min über eine Sauerstoffbrille) erhielt. Die Echokardiografie zeigte, dass Fabian vor allem eine zunehmende muskuläre, infundibuläre Enge (siehe Schema-Zeichnung Fallot-Tetralogie) entwickelt hatte, dass aber auch Pulmonalklappe und Pulmonal-arterie kaum gewachsen waren (Abbildung 2A und 2B). In dieser Konstellation entschieden wir uns für eine Operation mit Infundibulotomie und Kommissurotomie der bikuspiden Pulmonalklappe um die Lungendurchblutung zu verbessern und den Lungengefäßen bis zur Korrekturoperation noch einen weiteren Wachstumsreiz zu geben. Die Operation am Tag nach der stationären Aufnahme überstand Fabian gut. Er konnte am Folgetag der Operation von der Intensivstation verlegt werden und verließ eine Woche später mit stabilen Sauerstoffsättigungswerten von über 94 Prozent das Krankenhaus. Die Korrekturoperation kann nun geplant im Alter von sechs Monaten stattfinden.
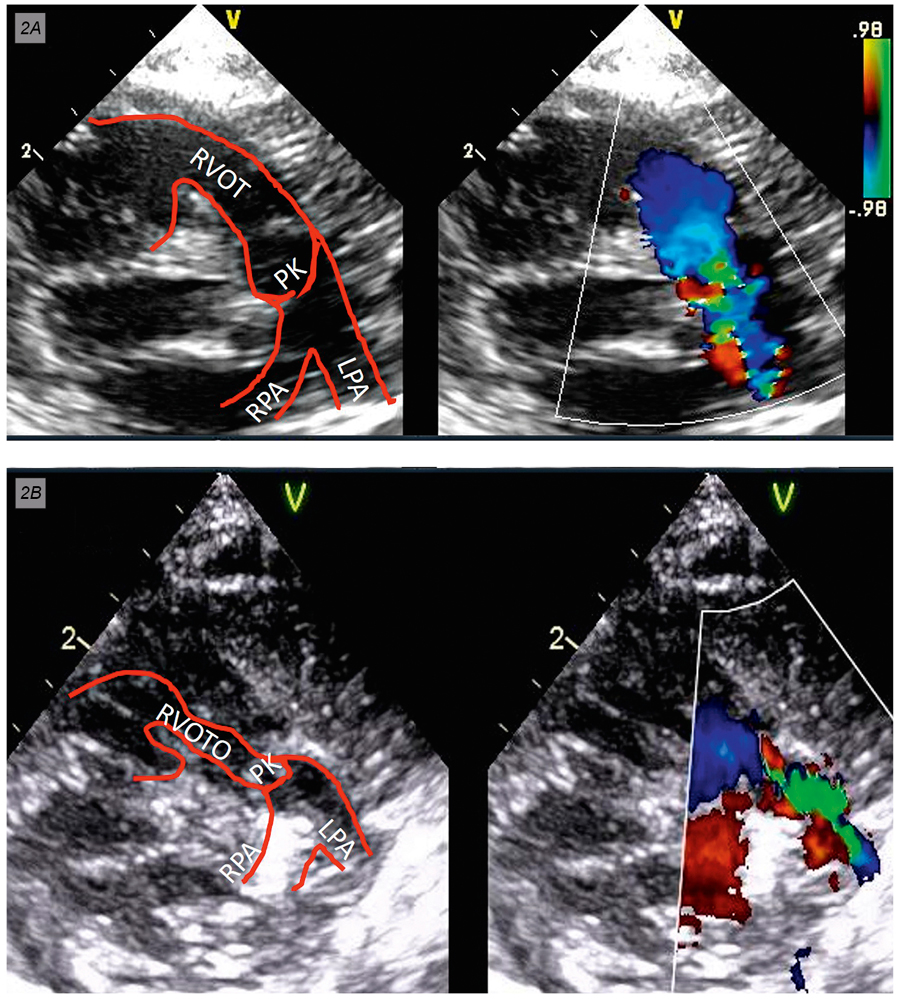
Abbildung 2: Darstellung des rechtsventrikulären Ausflusstraktes (RVOT) bei Fallot-Tetralogie.
A: am Tage der Geburt. B: im Alter von sechs Wochen. Die hypertrophierte Muskulatur führt zu einer langstreckigen muskulären Enge (RVOTO). LPA: linke Pulmonalarterie; PK: Pulmonalklappe; RPA: rechte Pulmonalarterie. Schmema-Zeichnung Fallot-Tetralogie: Je nach Ausmaß der Pulmonalstenose besteht bei der Fallot-Tetralogie ein mehr oder weniger ausgeprägter rechts-links Shunt, der zur Untersättigung in der Aorta führt. Ao: Aorta; PV: Pulmonalvenen; RV: Rechter Ventrikel; VCS: Vena cava superior; VCI: Vena cava inferior
Fallot-Tetralogie
Die Fallot-Tetralogie und die dextro-Transposition der großen Arterien (TGA) sind die beiden häufigsten zyanotischen angeborenen Herzfehler. Die Fallot-Tetralogie ist definiert als kombinierte Herzfehlbildung mit einem großen subaortalen Ventrikelseptumdefekt, mit einer dem Defekt „überreitenden Aorta“ (siehe Schema-Zeichnung Fallot-Tetralogie), einer Pulmonalstenose und einer rechtsventrikulären Hypertrophie. Der große, in der fetalen Echokardiografie gut sichtbare, Ventrikelseptumdefekt führt heute meist schon bei den pränatalen Screening-Untersuchungen zur Diagnose. Nach der Geburt kann der Herzfehler über das systolische Strömungsgeräusch der Pulmonalstenose leicht diagnostiziert werden. Viele Patienten mit Fallot-Tetralogie sind bei der Geburt (noch nicht) zyanotisch. Erst mit zunehmender Hypertrophie der Muskulatur des rechtsventrikulären Ausflusstraktes erhöht sich zunehmend der Widerstand, den der rechte Ventrikel für das Hineinpumpen des Blutes in die Lungenschlagader überwinden muss. Es entwickelt sich dann ein zunehmender Rechts-Links-Shunt (siehe Schema-Zeichnung Fallot-Tetralogie) über den Ventrikelseptumdefekt in die überreitende Aorta hinein. Die Korrekturoperation der Fallot-Tetralogie mit Verschluss des Ventrikelseptumdefektes und Beseitigung der rechtsventrikulären Ausflusstrakt-obstruktion findet regelhaft im Alter von vier bis sieben Monaten statt. Viele Patienten weisen bis dahin einen sehr stabilen Verlauf und ein gutes Gedeihen bei fehlender oder nur geringer stabiler Zyanose auf. Anders als bei einem Ventrikelseptumdefekt ohne rechtsventrikuläre Ausflusstraktobstruktion, verhindert diese Obstruktion die Lungenüberflutung und damit die Entwicklung einer klinischen Herzinsuffizienz. Patienten mit einer ausgeprägten muskulären Hypertrophie des rechtsventrikulären Ausflusstraktes und/oder mit einem elongierten muskulären rechtsventrikulären Ausflusstrakt können allerdings dynamische hochgradige rechtsventrikuläre Ausflusstraktobstruktionen entwickeln, sogenannte hypoxämische Anfälle. Die muskuläre dynamische Obstruktion lässt, in der Regel bei Aufregung und Tachykardie, nur noch eine so geringe Pulmonalperfusion zu, dass die Kinder kritisch entsättigen können, im Extremfall bis hin zu Ausbildung eines hypoxämischen cerebralen Krampfanfalles. In dieser Situation ist als Sofortmaßnahme die Sauerstoffgabe, gegebenenfalls eine intensivmedizinische Versorgung des Kindes mit Intubation und Beatmung, eine Sedierung und eine aortale Widerstandserhöhung, medikamentös, mit intravenöser Volumengabe oder durch Anpressen der Beine auf dem Körper angezeigt [6].
Wenn eine Korrekturoperation nach einem hypoxämischen Anfall noch nicht in Frage kommt, muss bis zum Erreichen des Korrekturalters die Lungendurchblutung gesichert werden. Hierfür kommen heute verschiedene operative und interventionelle Verfahren in Frage: operativ die Anlage eines aortopulmonalen Shunts oder eine partielle rechtsventrikuläre Ausflusstrakterweiterung, interventionell die Ballonvalvuloplastie der Pulmonalklappe oder die Einlage eines Stents in den rechtsventrikulären Ausflusstrakt. In seltenen Fällen kann bei Persistenz eines Ductus arteriosus auch ein Ductus-Stenting erfolgen [3].
Prognose nach Korrektur der Fallot-Tetralogie
Insbesondere bei Kindern mit Fallot-Tetralogie mit Pulmonalklappendysplasie und Hypoplasie nach ausgeprägter Infundibulektomie ist nach Korrekturoperation mit einer Pulmonalklappeninsuffizienz zu rechnen. Diese und die daraus möglicherweise entstehende rechtsventrikuläre Dilation stehen im Fokus der lebenslangen Nachsorge. Heute gibt es etablierte Leitlinienempfehlungen (AWMF online, Leitlinie „Pulmonalklappeninsuffizienz und Pulmonalklappenersatz im Kindes-, Jugend- und jungen Erwachsenenalter [EMAH-Patienten]“) für die Indikationsstellung zum Pulmonalklappenersatz nach Korrektur der Fallot-Tetralogie. Etwa 20 Prozent aller Patienten mit Fallot-Tetralogie benötigen im Laufe des Lebens nach der Korrekturoperation einen Pulmonalklappenersatz, meist erst in oder nach der Adoleszenz und meistens elektiv ohne ausgeprägte klinische Symptomatik oder Belastungseinschränkung bei zunehmender Dilatation des rechten Ventrikels [7].
Fall 3: Elektive Behandlung eines Vorhofseptumdefektes vom Sekundumtyp bei einem sechsjährigen Mädchen
Anamnese
Bei Sarah wurde der Vorhofseptumdefekt vom Sekundumtyp (ASD II) am Ende des ersten Lebensjahres bei der Vorsorgeuntersuchung U6 diagnostiziert. Es war ein systolisches Herzgeräusch auskultiert worden. Im Rahmen einer elektiven kinderkardiologischen Untersuchung wurde der ASD II dann echokardiografisch diagnostiziert. Da es Sarah immer gut ging und sie nie weniger belastbar und ausdauernd erschien als ihre zwei Jahre ältere Schwester, wurde das Vitium zunächst in jährlichen Abständen kinderkardiologisch kontrolliert. Gemäß Leitlinienempfehlungen wird Sarah nun elektiv im Vorschulalter zum Verschluss des ASD stationär eingewiesen. Bei einer Größe von 107 cm wiegt sie 15 kg was einem BMI von 13,1 kg/m2 entspricht. Damit hat Sarah leichtes Untergewicht, das möglicherweise kardial bedingt sein kann (siehe Tabelle Body-Mass-Index [BMI]).
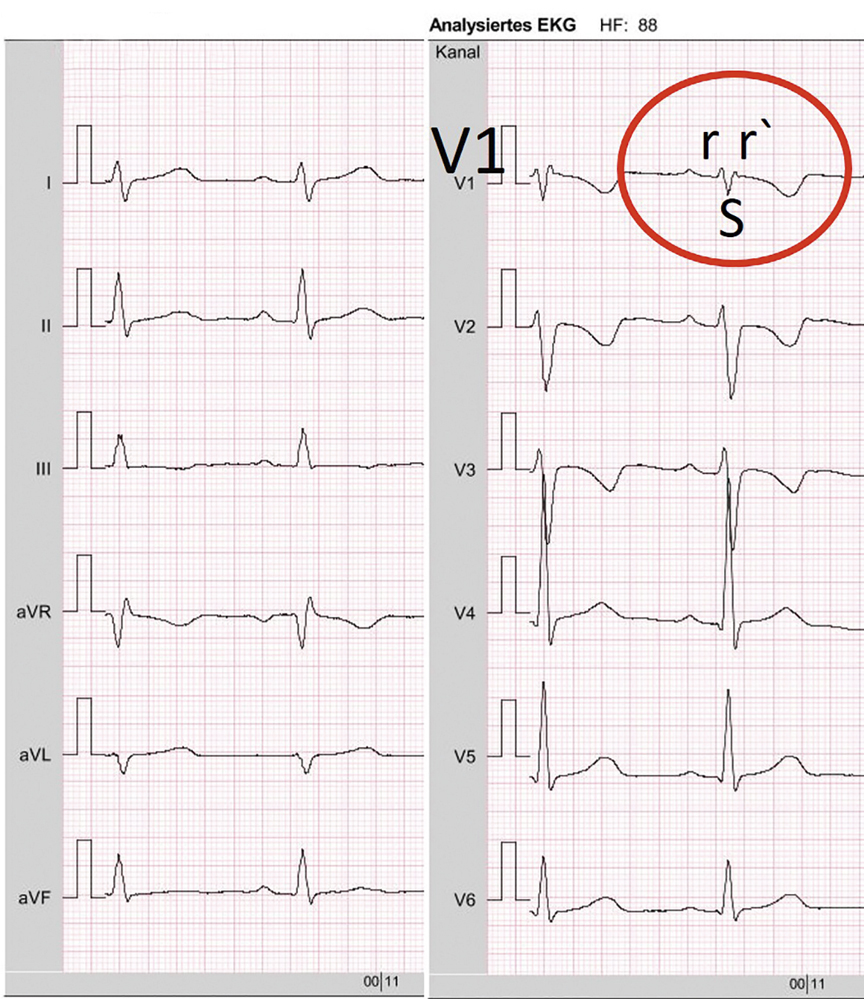
Abbildung 3: EKG einer sechsjährigen Patientin mit ASD II. Steiltyp, QRS-Dauer im Normalbereich. Etwas plumpe R´-Zacke in aVR. In den Brustwandableitungen inkompletter Rechtsschenkelblock rSr´-Form in V1. Etwas plumpe S-Zacke in V5 und V6. Das EKG entspricht einer Rechtsherzvolumenbelastung mit inkomplettem Rechtsschenkelblock.
Auskultatorisch besteht ein 2/6 Systolikum im zweiten ICR links parasternal und ein fixiert gespaltener 2. Herzton. Im EKG bestehen mit einem Steiltyp und einem inkompletten Rechtsschenkelblock Zeichen der Rechtsherzvolumenbelastung (Abbildung 3). Die Echokardiografie zeigt einen deutlichen vergrößerten rechten Vorhof und dilatierten rechten Ventrikel bei breitem Links-Rechts-Shunt über einem großen ASD II. Aufgrund der zentralen Lage erscheint der Defekt für den interventionellen Verschluss geeignet. Dieser erfolgt im Herzkatheterlabor unter Analgosedierung und transösophagealer Echokardiografie-Kontrolle. Während der Herzkatheteruntersuchung wird zunächst ein im Vorhofseptumdefekt liegender Ballonkatheter mit Röntgenkontrastmittel gefüllt (Abbildung 4A) um die genaue Defektgröße zu ermitteln. Im Anschluss kann der Defekt mit einem passenden Amplatzer ASD Occluder verschlossen werden (Abbildung 4B-4D). Sarah erhält für sechs Monate eine Medikation mit Low-Dose-Acetylsalicylsäure (3-5 mg/kg/Tag). Sie kann am Folgetag nach Implantation des Schirmes aus der stationären Behandlung entlassen werden. Eine erste echokardiografische Kontrolluntersuchung erfolgt kurzfristig nach einer Woche und zeigt das komplikationslose Einheilen des Schirmes.
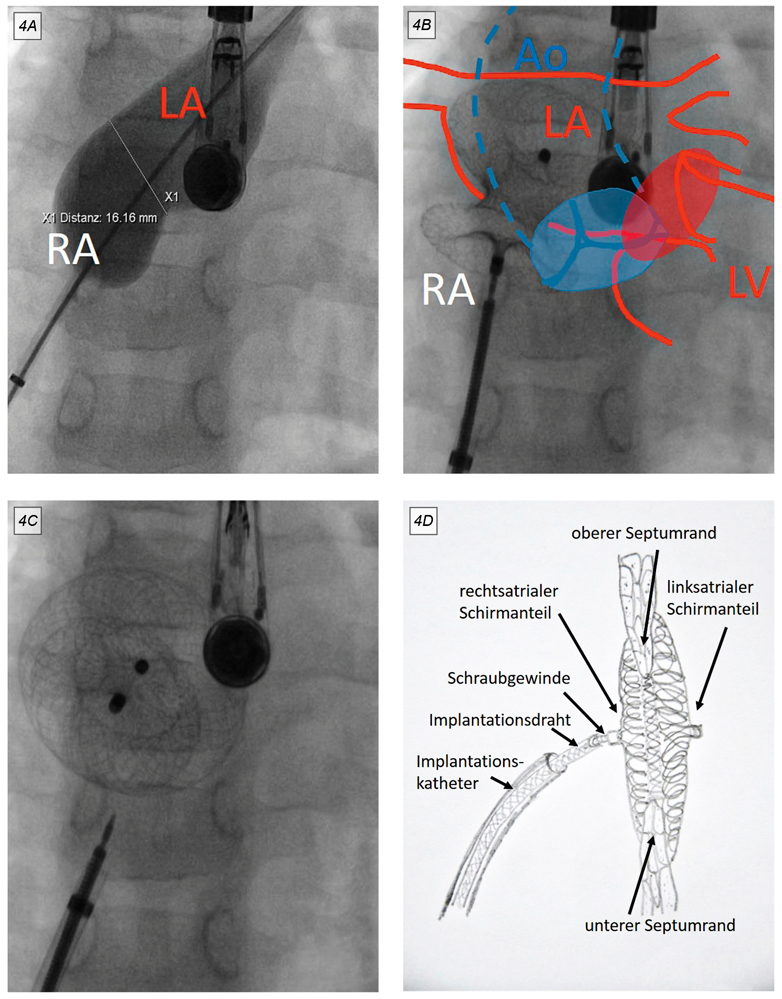
Abbildung 4: Interventioneller ASD-Verschluss mit einem Amplatzer ASD-Occluder in der pa-Projektion der Röntgenröhre. Die Herzkatheter gelangen über die Femoralvene in den rechten Vorhof (RA) und über den Vorhofseptumdefekt in den linken Vorhof (LA). A: Ausmessen der Defektgröße mit einem im Defekt mit Röntgenkontrastmittel gefüllten Ballonkatheter. B: am linksseitigem Vorhofseptumrand aufgespannter Schirm. Schematisch dargestellt sind die in der Fluoroskopie nicht sichtbaren Landmarken: der dorsal liegende linke Vorhof (LA) und die Mitralklappe (roter Kreis), ventral die Aortenklappe (blauer Kreis) und die Aorta (Ao), rechts ventral der rechte Vorhof (RA). Auf allen Aufnahmen sichtbar ist die im Ösophagus liegende Echokardiografie-sonde zur Steuerung der Implantation. C: abgelöster und flach konfigurierter Schirm. D: Die schematische Darstellung zeigt die Verankerung des ASD-Verschlussschirmes im Vorhofseptumdefekt.
Vorhofseptumdefekt vom Sekundumtyp
Ein behandlungsbedürftiger ASD II ist die häufigste elektive Einweisungsdiagnose zur Korrektur eines angeborenen Herzfehlers. Einziges frühes Symptom ist oft das leise systolische Herzgeräusch und die fixierte Spaltung des zweiten Herztones bei der Auskultation. Bei einem Verschluss eines ASD II im Vorschulalter ist eine Normalisierung der rechtsatrialen und rechtsventrikulären Dilatation durch die Volumenbelastung zu erwarten. Langzeitfolgen bei einem nicht diagnostizierten oder nicht behandelten ASD II können Vorhofarrhythmien,
pulmonale Symptome aufgrund der fortbestehenden Volumenbelastung, im späterem Lebensalter auch Rechtsherzinsuffizienz und in seltenen Fällen pulmonal-arterielle Hypertension sein. Ebenfalls selten führen thrombembolische cerebrale Ischämien zur Spätdiagnose übersehener Vorhofseptumdefekte.
Vorhofseptumdefekte können mit verschiedenen Schirmen transvenös verschlossen werden, wenn ausreichende Randstrukturen vorhanden sind. Der operative Verschluss, entweder durch direkte Naht, mit einem Flicken aus Eigenperikard oder mit anderen Patch-Materialien ist ebenfalls in der Routine sicher und komplikationsarm durchzuführen [8]. Neben der medianen Thorakotomie gibt es für den Verschluss des Vorhofseptumdefektes minimalinvasivere Zugangswege und Operationsmethoden.
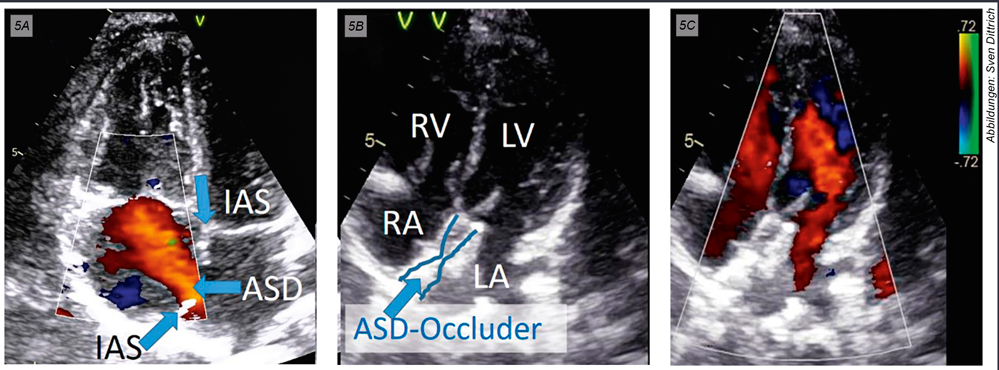
Abbildung 5: Die Pfeile in Abbildung 5A demonstrieren Teile des superioren und inferioren Vorhofseptums und den mittigen Defekt; ASD: atrialer Septumdefekt; IAS: interatriales Septum. Darstellung des Vorhofseptumdefektes in der transthorakalen Echokardiografie links (A) vor und rechts (B und C) nach Implantation des ASD-Occluders. LA: linker Vorhof; LV: linker Ventrikel; RA: rechter Vorhof; RV: rechter Ventrikel.
Nach ASD-Verschluss ist die Langzeitprognose ausgezeichnet [9].
Das Literaturverzeichnis kann im Internet unter www.bayerisches-aerzteblatt.de (Aktuelles Heft) abgerufen werden.
Die Autoren erklären, dass sie keine finanziellen oder persönlichen Beziehungen zu Dritten haben, deren Interessen vom Manuskript positiv oder negativ betroffen sein könnten.
Das Wichtigste in Kürze
Die drei Fälle zeigen exemplarisch das Spektrum der Diagnostik und der Primärbehandlung angeborener Herzfehler.
Bei Neugeborenen sind ductusabhängige Herzfehler wichtige Differenzialdiagnosen mit Zyanose oder mit kardiorespiratorischer Dekompensation einhergehender akuter schwerer Krankheitsbilder. Die meisten angeborenen Herzfehlbildungen werden nach früher Diagnosestellung im Neugeborenen- oder Säuglingsalter elektiv behandelt und bedürfen bis dahin einer intensivierten Überwachung und Betreuung um mögliche kritische Komplikationen des noch nicht korrigierten Herzfehlers rechtzeitig zu erkennen.
Zu den wichtigsten Komplikationen gehören hier die Zyanose und die Herzinsuffizienz bei Shuntvitien. Patientinnen und Patienten mit einem Vorhofseptumdefekt gehören in das Routinespektrum der Diagnostik und Therapie angeborener Herzfehler. Der Verschluss des Defektes normalisiert die Hämodynamik und ein zeitiger gelungener Verschluss eines Shuntvitiums verspricht eine exzellente Langzeitprognose.
Bei komplexeren Herzfehlern wie der Fallot-Tetralogie wird die Langzeitprognose möglicherweise durch fortbestehende hämodynamische Belastungen beeinflusst, zum Beispiel durch eine Pulmonalklappeninsuffizienz.
Für die meisten angeborenen Herzfehler sind daher nach Korrektur lebenslang regelmäßige kardiologische Kontrollen im Sinne einer Sekundärprävention empfohlen.
Autoren

Professor Dr. Sven Dittrich 1

Dr. med. Dr. rer. biol. hum. Isabelle Schöffl 1

Dr. Wolfgang Wällisch 1

Professor Dr. Robert Cesnjevar 2
Kinderherzzentrum im Universitätsklinikum Erlangen, Loschgestraße 15, 91054 Erlangen
1 Kinderkardiologische Abteilung
2 Kinderherzchirurgische Abteilung
Teilen:
Das könnte Sie auch interessieren: