In der Europäischen Union (EU) gilt eine Erkrankung als selten, wenn nicht mehr als fünf von 10.000 Menschen in der EU von ihr betroffen sind. Das „Bayerische Ärzteblatt“ greift in der Serie „Seltene Erkrankungen“ sowohl methodische und systematische Aspekte auf und berichtet auch über einzelne seltene Erkrankungen (SE). Ziel ist es, durch die verschiedenen Beiträge, die Befassung mit diesem heterogenen Thema anzuregen und eine Sensibilisierung zu erreichen.
05.10.2020
Varia
![]() Artikel als PDF
Artikel als PDF
Kawasaki-Syndrom
 Kawasaki-Syndrom
Kawasaki-Syndrom
Eine akute, systemische und selbst begrenzende fieberhafte Erkrankung mit nekrotisierender Vaskulitis hauptsächlich kleiner und mittlerer Arterien
Im vierten Teil der Serie schreiben Professorin Dr. Johannes-Peter Haas und Dr. Toni Hospach über das Kawasaki-Syndrom (KS).
Das KS ist eine hochfieberhafte Vaskulitis des Kleinkindes unbekannter Ursache. Bei zumeist guter Prognose verhindert eine frühzeitige Therapie die Entstehung von Koronararterienaneurysmen. Seit Mai 2020 wird ein Kawasaki-ähnliches Syndrom (MIS-C) assoziiert mit COVID-19 berichtet.
Die Erkrankung wurde zuerst 1967 von Tomisaku Kawasaki als „acute febrile mucocutaneous lymph node syndrome“ beschrieben [1]. Das KS ist bei Kindern japanischer Abstammung am häufigsten (Inzidenz 2015/2016 330/100.000). Auch in den USA tritt das KS am häufigsten bei Kindern asiatischer Abstammung (Hospitalisierung 39/100.000 bei Kindern unter fünf Jahren) und am seltensten bei Kindern kaukasischer Herkunft (ca. 11/100.000) auf. Typischerweise erkranken Kleinkinder (75 Prozent der Patienten sind unter fünf Jahre) mit Gipfel im zweiten Lebensjahr. Jungen sind etwas häufiger betroffen als Mädchen (ca. 1,5 zu 1).
In den Wintermonaten wird ein deutlicher Anstieg der Erkrankungsfrequenz beobachtet [2, 3, 4].
Am COVID-19 assoziierten multisystemic inflammatory syndrome (MIS-C – siehe Kasten S. 491), einer KS-ähnlichen Erkrankung erkranken auch Jugendliche und Erwachsene [5].
Ätiologie und Pathogenese
Die Ursache ist unbekannt und Hypothesen zur Ätiologie und Pathogenese des KS widersprechen sich teilweise [6]. Viele Befunde sprechen für eine infektiöse Ursache: Kinder unter drei Monaten erkranken sehr selten an KS (möglicherweise durch den Schutz durch maternale Antikörper). Auch die saisonal abhängige Inzidenz und das Auftreten von Epidemien unterstützen die Infektionshypothese ebenso wie der üblicherweise selbstlimitierende Verlauf der Erkrankung. Kawasaki-ähnliche Erkrankungen wie das MIS-C wurden eindeutig mit bestimmten viralen Infektionen assoziiert beobachtet.
Das KS ähnelt manchen hochfieberhaften exanthematischen Infektionserkrankungen. Trotz dieser Ähnlichkeiten konnte ein Keimnachweis bisher nicht geführt werden, wobei viele infektiöse Agenzien untersucht wurden; unter anderem: Retroviren, Adenoviren, Epstein-Barr-Virus (EBV), Humanes Herpesvirus 6, Chlamydien, Streptokokken, Mykobakterien und Hausstaubmilben.
Unabhängig von den auslösenden Ursachen wird allgemein eine genetische Prädisposition vermutet, wofür auch die höhere Inzidenz der Erkrankung bei japanischen Kindern spricht, die auch nach einem Umzug zum Beispiel in die Vereinigten Staaten erhöht bleibt. Genomweite Assoziationsstudien wiesen eine Reihe von Kandidatengenen nach, die mit dem Risiko, an einem KS zu erkranken, bzw. mit dem Risiko einer kardiovaskulären Beteiligung oder aber mit einer Resistenz gegenüber einer Immunglobulintherapie assoziiert sind [7, 8].
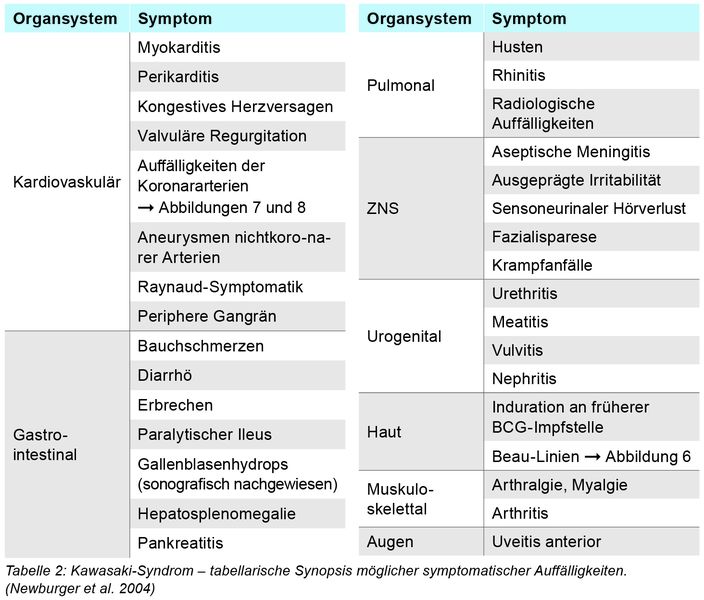
Das KS ist eine generalisierte Vaskulitis mit einer Prädilektion für die mittelgroßen Arterien, obwohl Gefäße aller Größenordnungen betroffen sein können. Zusätzlich ist eine systemische Entzündung in vielen Organen vorhanden, wie zum Beispiel Herz (Myokarditis), Nervensystem (aseptische Meningitis), Lunge (Pneumonitis), Magen-Darm-Trakt (Stomatitis, Enteritis, Hepatitis), Urogenitalsystem (Nephritis, Zystitis) und dem hämatopoetischen System (Lymphadenitis). Die entsprechende Aktivierung des Immunsystems schließt das zelluläre System, proinflammatorische Zytokine, Endothelzellaktivierung, Elastasen, Metalloproteinasen und Wachstumsfaktoren ein.
Klinische Symptome
Die Manifestation der vielfältigen Symptome (siehe Tabelle 1 und 2 und Abbildungen 1 bis 8) beim KS variiert im Verlauf der Erkrankung. Es werden drei Phasen unterschieden:
Akute febrile, inflammatorische Periode -(ein bis zwei Wochen Dauer): Meist beginnt das Fieber abrupt und es entwickeln sich im Verlauf von drei bis vier Tagen typische Symptome (Tabelle 1). Die Diagnose muss nach Möglichkeit in dieser Phase gestellt werden.

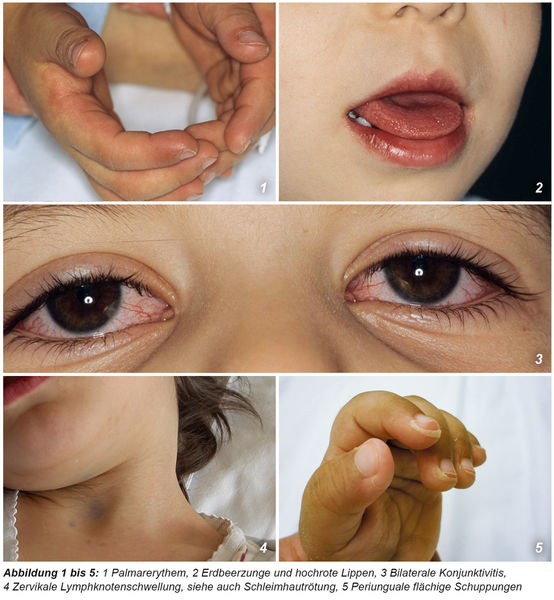
Subakute Phase (zwei bis vier Wochen Dauer): Unbehandelt entfiebern die Patienten in Woche drei bis vier. Typisch ist in diesem Zeitraum eine Hautschuppung an Händen und Füßen. Bei Kindern nach Immunglobulintherapie sind in dieser Phase häufig bis auf die Schuppung keine Symptome mehr vorhanden.
Phase der Rekonvaleszenz, die Monate dauern kann. Die meisten Kinder sind während dieser Phase ohne Symptome, gelegentlich besteht aber noch Müdigkeit und Leistungsschwäche.
Typisch ist, dass die genannten Symptome sequenziell auftreten können, was die Diagnosesicherung erschweren kann. Bei Vorliegen von ≥ vier der klassischen Symptome kann die Diagnose KS auch schon nach vier Tagen Fieber gestellt werden kann. Die Vielfalt der möglichen Begleitsymptome ist sehr umfänglich (Tabelle 2) und kann die Diagnostik erheblich erschweren.
Diagnose
Die Diagnose KS kann nicht anhand pathognomonischer klinischer Befunde gestellt werden, sondern ist abhängig von diagnostischen Kriterien (Tabelle 1). Neben der klinischen Beobachtung kommt der genauen Anamnese eine besondere Bedeutung zu, da die Symptome auch sequenziell auftreten können. Kinder, die die in Tabelle 1 genannten Kriterien nicht erfüllen, werden als Kinder mit einem „atypischen“ oder „inkompletten“ KS diagnostiziert. Dabei sollte der Begriff „atypisch“ für Krankheitsverläufe reserviert sein, die sich mit sehr ungewöhnlichen und seltenen Symptomen präsentieren [9].
Es gibt keine beweisenden oder spezifischen Labor-untersuchungen für das KS. Die Labordiagnostik ist zum Ausschluss anderer Differenzialdiagnosen und bei der Abklärung eines „inkompletten KS“ von großer Bedeutung. Ein CRP-Wert ≥ 3 mg/dl und eine Blutsenkungsgeschwindigkeit (BSG) ≥ 40 mm n.W./h unterstützen die Verdachts-diagnose ebenso wie die folgenden Laborparameter nach ≥ 5 Tagen Fieber: Albumin ≤ 3 g/dl, Anämie, Glutamat-Pyruvat-Transaminase (GPT) erhöht, Thrombozyten ≥ 450.000 am Tag 7, Leukozyten ≥ 15.000/µl und im Urin 10 Leukozyten/Gesichtsfeld. Für die differenzialdiagnostische Abklärung sind Blutkulturen, Antistreptokokkentiter, Rachenabstrich, Stuhlkulturen, Leber- und Nierenfunktionswerte inklusive Gerinnung, ANA-Screening und serologische Untersuchungen sinnvoll.
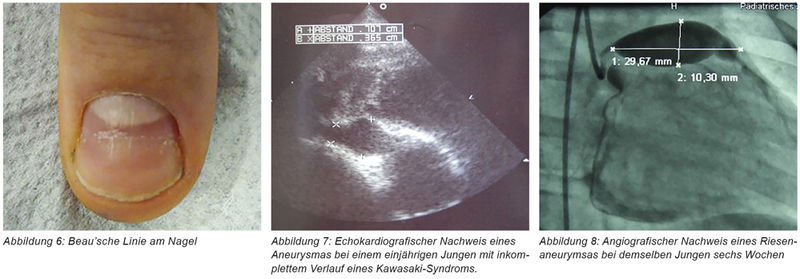
Die Durchführung einer 2D-Echokardiografie durch einen Kinderkardiologen ist bereits beim Verdacht eines KS angezeigt und sollte bei unkompliziertem Verlauf nach zwei und sechs bis acht Wochen wiederholt werden. Weiteres Monitoring ist bei Kindern mit nachgewiesener Beteiligung der Koronararterien indiziert. Da die normale Weite der Koronararterien abhängig von der Körperoberfläche ist, sollte dies bei den Messungen berücksichtigt werden [2]. Im EKG finden sich eventuell Arrhythmie, ein verlängertes PR-Intervall oder unspezifische ST- und T-Wellen-Veränderungen. Schwere Verläufe können sich mit den Zeichen eines kardiogenen Schocks präsentieren.
Differenzialdiagnose
Differenzialdiagnostisch müssen virale Erkrankungen (unter anderem Adenoviren, Röteln, Masern, Enteroviren, EBV, Parvoviren, Cytomegalovirus [CMV]) abgeklärt werden. Dabei ist zu bedenken, dass der Nachweis einer viralen Erkrankung des Respirationstrakts (zum Beispiel RS-Viren, Metapneumovirus, Coronavirus, Parainfluenzavirus oder Influenzavirus) die Diagnose KS nicht ausschließt. Ein Drittel der Kinder mit typischem KS wiesen zeitgleich Infektionen des Respirationstraktes auf [10]. Außerdem sind bakterielle Erkrankungen wie Scharlach, toxisches Schocksyndrom, das „staphylococcal scalded skin syndrome“, Leptospiren unter anderem auszuschließen. Vor allem autoinflammatorische Erkrankungen wie die systemische Form der juvenilen idiopathischen Arthritis und Vaskulitiden wie die Polyarteritis nodosa können ein KS oder ein inkomplettes KS imitieren. Ebenfalls sollten maligne Erkrankungen (Leukämie, Histiozytose) und unerwünschte Arzneimittelwirkungen (drug-fever) bedacht werden.
Therapie
Die Therapie hat die Reduktion der Entzündung und die Vermeidung von Koronararterienaneurysmen zum Ziel. Da diese meist in der zweiten bis dritten Krankheitswoche entstehen, muss die Behandlung so früh wie möglich, spätestens bis zum zehnten Krankheitstag, begonnen werden.
Die kombinierte Gabe von intravenösen Immunglobulinen und von Acetylsalicylsäure stellt die etablierte, evidenzbasierte Behandlung in der ersten Krankheitsphase bei unkomplizierten Verläufen dar. Die Einzelgabe von 2 g/kg Intravenöser Immunglobuline (IVIG), stellt die Standardtherapie des KS dar. Auch bei einer verzögerten Diagnosestellung scheint noch ein therapeutischer Nutzen gegeben zu sein. Die IVIG-Infusion wird unter Monitorüberwachung verabreicht. Bei Risikopatienten lässt sich das Auftreten von Koronaraneurysmen durch die zusätzliche Gabe von Steroiden signifikant senken [11, 12]. Bei Nichtansprechen auf diese Therapie oder bei Auftreten von Komplikationen (Rezidiv, Aneurysmen) kommen auch Interleukin-1-Inhibitoren oder TNF-Blocker zum Einsatz.
Patienten mit einem hohen Risiko einer Koronararterienerkrankung oder einem möglichen Nichtansprechen auf die Standard-IVIG-Therapie sollten schnell und sicher identifiziert werden, mit der Konsequenz einer erweiterten Therapie. Die bislang publizierten Scores zur Risikobewertung zeigten gute Sensitivitäts- und Spezifitätsergebnisse bei japanischen Patienten und weniger klare Trennschärfe bei anderen Ethnien (zum Beispiel Deutschland, Italien [13, 14]). Damit bleibt die Identifizierung von nicht-asiatischen Patienten, die möglicherweise nicht auf IVIG ansprechen und zur Vermeidung von Koronararterienaneurysmen (KAA) weitere Therapien benötigen, weiter eine Herausforderung [15].
Der Einsatz einer adjuvanten mehrtägigen Steroidtherapie (2 mg/kg Prednisolon) in der Initialphase des KS ist für Hochrisikopatienten mit einer echokardiografisch nachgewiesenen Erweiterung der Koronararterien ab einem Z-Score von ≥ 2, einem Erkrankungsalter im ersten Lebensjahr, einer initialen Schocksymptomatik und einem Makrophagenaktivierungssyndrom zu empfehlen. Bei weiteren – weniger ausgeprägten – Risikofaktoren wie deutlich erhöhten Inflammationsparametern, erhöhten Leberwerten, Anämie, Hypalbuminämie, Hyponatriämie, männlichem Geschlecht, Alter > sieben Jahre, einer Krankheitsdauer < = vier oder > 14 Tagen ist eine solche Steroidtherapie zu erwägen [16 bis 21].
Eine initiale Therapie mit TNF-Blockade ist nach dem aktuellen Wissensstand nicht indiziert. Infliximab ist jedoch eine Therapieoption bei IVIG- und steroidresistenten Krankheitsverläufen [22 bis 26]. Nach bisherigen Studienergebnissen kann ein frühzeitiger Einsatz von Ciclosporin A (CSA) bei Risikopatienten in der Primärtherapie des KS sinnvoll sein, es sollten aber noch weitere Studienergebnisse vorliegen, bevor ein allgemeiner Einsatz empfohlen werden kann. Bei IVIG-refraktären Patienten sollte CSA aufgrund der limitierten Studienergebnisse nachrangig eingesetzt werden [27].
Initial IVIG-resistente Krankheitsverläufe
Ungefähr 10 bis 20 Prozent aller Patienten mit KS sprechen auf die initiale Therapie mit IVIG und ASS nicht an, die Patienten fiebern ≥ 36 Stunden nach Ende der initialen IVIG-Infusion immer noch oder wieder [28]. IVIG-refraktäre Patienten haben ein erhöhtes Risiko für Koronaraneurysmen. Neben einer erneuten IVIG-Therapie (2 g/kg KG) sollten in dieser Situation frühzeitig zusätzliche antiinflammatorische Therapien in Erwägung gezogen werden, um das Risiko der Ausbildung von KAA zu minimieren [29 bis 32]. Die Wirksamkeit einer Pulssteroidtherapie bei IVIG-resistentem KS wurde von Hashino et al. untersucht: 17 von 262 Patienten, die nicht auf eine erste IVIG-Einzelinfusion (2 g/kg) und ASS (30 mg/kg/Tag) und auch nicht auf eine erneute IVIG-Behandlung (1 g/kg) angesprochen hatten, wurden randomisiert entweder mit einer dritten IVIG-Therapie (1 g/kg) oder mit 20 mg/kg Methylprednisolon behandelt. Die Inzidenz von Koronararterienveränderungen war bei beiden Gruppen hoch (60 bis 75 Prozent), aber nicht signifikant unterschiedlich. Bei den mit Methylprednisolon behandelten Kindern war die Fieberdauer signifikant kürzer [33].
Interleukin-1-Rezeptorantagonist (Anakinra): Nach ersten positiven Fallberichten [34] wurden im „Kawakinra trial“ 16 IVIG-refraktäre Patienten eingeschlossen. Eine 15-tägige Behandlung mit Anakinra in einer Dosierung von 2 bis 6 mg/kg war hocheffektiv im Hinblick auf die Inflammation und Symptomatik [35]. Eine weitere Studie zum Einsatz von Anakinra bei erweiterten Koronarien (ANAKID trial) soll Ende 2020 abgeschlossen werden [36].
Langzeitüberwachung und Prognose
Die Dauer der notwendigen Überwachung nach einem KS mit fehlender oder geringer transienter Dilation der Koronararterien in der akuten Phase wird unterschiedlich diskutiert. Aktuell wird von der „American Heart Association“ empfohlen, diese Überwachungsmaßnahmen auf ein Jahr nach Erkrankungsbeginn zu begrenzen [2, 19, 37].
Die Prognose des KS in Hinsicht auf die Lebensqualität wird im Wesentlichen durch das Auftreten von Koronorarterienaneurysmen bestimmt. Die Mortalität aller hospitalisierten Kinder wird mit 0,12 bis 0,17 Prozent angeben, sie liegt aber bei Kindern über zehn Jahren deutlich höher. Bei Patienten mit sehr großen Aneurysmen liegt die Überlebensrate zehn Jahre nach Erkrankungsbeginn bei 95 Prozent und sinkt nach weiteren 20 Jahren auf 88 Prozent ab. Auch bei Patienten, bei denen ein Koronararterien-Bypass notwendig geworden war, lag die Überlebensrate nach 25 Jahren noch bei 95 Prozent [23, 38 bis 40].
Das Literaturverzeichnis kann im Internet unter www.bayerisches-aerzteblatt.de (Aktuelles Heft) abgerufen werden.

Autoren
Professor Dr. Johannes-Peter Haas 1
Dr. Toni Hospach 2
1 Geschäftsführender Ärztlicher Direktor, Deutsches Zentrum für Kinder- und Jugendrheumatologie, Garmisch-Partenkirchen, Gehfeldstr. 24, 82467 Garmisch-Partenkirchen
2 Leiter des Zentrums für pädiatrische Rheumatologie am Klinikum Stuttgart, Kriegsbergstr. 62, 70174 Stuttgart
Teilen:
Das könnte Sie auch interessieren: