16.01.2020
Varia
![]() Artikel als PDF
Artikel als PDF
Angeborene Störungen der Immunität ‒ Seltene Erkrankungen in der Kinder- und Jugendmedizin
 © Andrea Haase / mauritius-images.com
© Andrea Haase / mauritius-images.com
In der Europäischen Union (EU) gilt eine Erkrankung als selten, wenn nicht mehr als fünf von 10.000 Menschen in der EU von ihr betroffen sind. Das „Bayerische Ärzteblatt“ greift in der Serie „Seltene Erkrankungen“ sowohl methodische und systematische Aspekte auf und berichtet auch über einzelne seltene Erkrangungen (SE). Ziel ist es, durch die verschiedenen Beiträge, die Befassung mit diesem heterogenen Thema anzuregen und eine Sensibilisierung zu erreichen.
Den zweiten Teil der Serie schreiben Privatdozent Dr. Dr. sci. nat. Fabian Hauck et al. über seltene Erkrankungen in der Kinder- und Jugendmedizin am Beispiel angeborener Störungen der Immunität.
SE sind in der EU durch ihre Prävalenz definiert: wenn weniger als fünf Menschen von 10.000 an einer Erkrankung leiden, so gilt sie als selten [1]. Gegenwärtig sind über 7.000 SE bekannt. Ihre Zahl steigt stetig an, und konservative Schätzungen gehen von einer Gesamtzahl von weit über 8.000 aus. Annähernd fünf Prozent der Weltbevölkerung, also etwa 350 Millionen Menschen, leben mit einer SE. In Deutschland geht man von ca. vier Millionen Patienten aus. Die meisten SE haben eine genetische Ätiologie und manifestieren sich bereits im Kindes- und Jugendalter. Etwa 80 Prozent der SE verteilen sich auf die häufigsten vier Prozent der Entitäten mit einer für SE relativ hohen Prävalenz von 1 bis 5:10.000 [2].
Zum Zeitpunkt der ärztlichen Approbation kennt ein angehender Arzt im Mittel 2.000 bis 2.500 überwiegend häufige Symptome und Erkrankungen insbesondere aus den medizinischen Fachgebieten der Inneren Medizin, der Neurologie und der Chirurgie [3]. Auch wenn sich die Zeit bis zum Stellen einer korrekten Diagnose im Laufe der vergangenen Jahre verkürzt hat, so dauert es immer noch viel zu lange, bis Patienten Klarheit über ihre Erkrankung erhalten. Durchschnittlich werden acht verschiedene Ärzte aufgesucht, von der klinischen Manifestation bis zur ärztlichen Diagnosestellung vergehen durchschnittlich fünf Jahre. Zahlreiche Menschen mit SE bekommen (zunächst) eine Fehldiagnose und/oder Fehlbehandlung oder bleiben trotz hoher Morbidität und Mortalität gänzlich ohne Diagnose und Behandlung [4]. Häufig gelingt es nicht, die für eine gute interdisziplinäre medizinische Versorgung nötige ärztliche Expertise an etablierten Zentren aufzubauen und vorzuhalten. Auch die medizinischen Daten, der in der Regel dezentral betreuten Patienten, werden kaum zusammengeführt [5].
Internationale Ebene
SE als Krankheitsgruppe haben in den vergangenen Jahren auf internationaler Ebene den abstrakten Status eines globalen Problems der öffentlichen Gesundheitspflege erlangt. Verschiedene Initiativen zur Verbesserung der Gesundheitsfürsorge von Menschen mit SE wurden vorgeschlagen oder konkret initiiert [6]. Auf europäischer Ebene fördert die Europäische Kommission die Einrichtung Europäischer Referenznetze für komplexe und niedrig prävalente Erkrankungen (European Reference Networks, ERN) [7], das Europäische Gemeinsame Programm für Seltene Erkrankungen (European Joint Programme on Rare Diseases, EJP RD) [8] und ein spezialisiertes Register in der Europäischen Infrastruktur zur Registrierung Seltener Erkrankungen (European Rare Disease Registry Infrastructure, ERDRI) [9].
Auf nationaler Ebene haben das Bundesministerium für Bildung und Forschung (BMBF), das Bundesministerium für Gesundheit (BMG) und der Verein Allianz Chronischer Seltener Erkrankungen (ACHSE e. V.) im Jahr 2010 das Nationale Aktionsbündnis für Menschen mit Seltenen Erkrankungen (NAMSE) ins Leben gerufen [10]. Dieses Aktionsbündnis arbeitet mit insgesamt 28 Gremien und Organisationen des deutschen Gesundheitswesens zusammen und veröffentlichte im Jahr 2013 den Nationalen Aktionsplan für Menschen mit Seltenen Erkrankungen [11]. Darin sind Handlungsfelder, Empfehlungen und 52 Maßnahmenvorschläge zur Verbesserung der Versorgung von Menschen mit SE beschrieben. Insbesondere die Einrichtung von Zentren für Seltene Erkrankungen (ZSE), die Entwicklung interoperabler digitaler Strukturen, die Erfassung der SE mit der sogenannten ORPHA-Kodierung (Orphacode) [12] und eine bedarfsgerechte Leistungsvergütung sollen eine qualitativ hochwertige Versorgung von Menschen mit SE ermöglichen und die medizinische Forschung zu Grundlagen, Mechanismen und Behandlungen von SE fördern.
Genetische Natur
Aufgrund der überwiegend genetischen Natur der SE und der im Vergleich zur Erwachsenenmedizin überdurchschnittlichen Prävalenz im Patientenkollektiv der Kinder- und Jugendmedizin, befasst sich die Pädiatrie seit jeher und aus ihrem Wesen heraus mit SE. Gerade weil die nachhaltige Versorgung von Kindern und Jugendlichen mit SE national nur sektorenübergreifend und interdisziplinär gelingen kann, steht insbesondere die universitäre Kinder- und Jugendmedizin vor großen strukturellen und inhaltlichen Herausforderungen. Einerseits kommt ihr sui generis von jeher eine gewisse Vorreiterrolle in der Ausgestaltung dieser Themenfelder zu [13], andererseits sieht sie sich in einer Zeit, in der sich ein Gesundheitswesen immer mehr durch Prinzipien der Ökonomie durchdringen lässt, vor enormen Herausforderungen.
Care-for-Rare Center
An der Kinderklinik und Kinderpoliklinik im Dr. von Haunerschen Kinderspital am Klinikum der Universität der Ludwig-Maximilians-Universität (LMU) München sind daher im Kontext der oben genannten Initiativen mehrere Verbünde und Strukturen entstanden. Am Münchner Zentrum für Seltene Erkrankungen (MZSE) [14] werden im Sinne eines NAMSE-Typ-A-Zentrums grundsätzlich alle Menschen mit SE gelenkt. Die aktuellen Schwerpunkte des MZSE im Bereich der Erwachsenenmedizin liegen auf neuromuskulären, dermatologischen, immunologischen und kardiologischen Erkrankungen. Kinder und jugendliche Patienten werden am Care-for-Rare Center (C4RC) [15] des Dr. von Haunerschen Kinderspitals betreut.
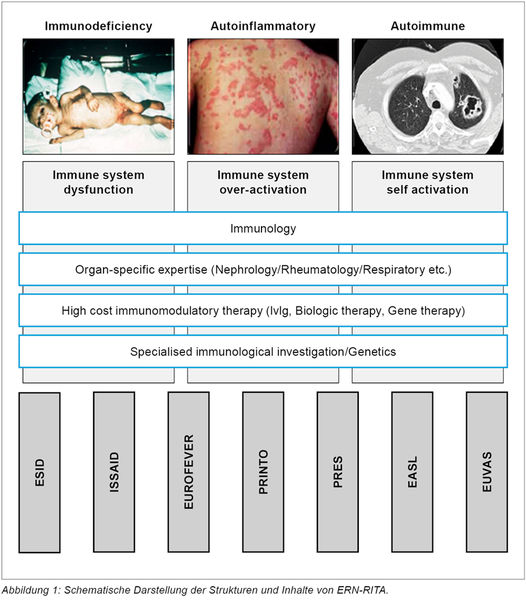
Eines der zahlreichen NAMSE-Typ-B-Zentren am C4RC befasst sich exemplarisch mit Kindern und Jugendlichen mit angeborenen Störungen der Immunität (inborn errors of immunity). Strukturell ist das Klinikum der LMU über die Abteilung für Pädiatrische Immunologie und Rheumatologie auf europäischer Ebene im European Reference Network for Rare Primary Immunodeficiency, Autoinflammatory and Autoimmune Diseases (ERN-RITA) eingegliedert. Hier werden auch die Aspekte zur Versorgung von Kindern mit Immundefekterkrankungen koordiniert (Abbildung 1) [16]. Das primäre Ziel von ERN-RITA ist eine europäische, grenzüberschreitende Versorgung von Menschen mit seltenen Immunerkrankungen, um unabhängig der Nationalitätenzugehörigkeit und der nationalen medizinischen Gegebenheiten einen Zugang zur bestmöglichen Versorgung zu gewährleisten. Dies geschieht konkret durch eine digitale Plattform, das sogenannte „Clinical Patient Management System“ (CPMS) [17], über das datenschutzkonform virtuelle Expertenpanels zusammengestellt werden, um komplexe und niedrig prävalente Patientenfälle mit seltenen Immunerkrankungen zu diskutieren. Zusätzlich werden im ERN-RITA europäische Leitlinien zur Diagnostik und Therapie von Menschen mit angeborener Störung der Immunität konsentiert und im Internet barrierefrei publiziert [16]. ERN-RITA integriert neben der ärztlichen Kompetenz aktiv die relevanten wissenschaftlichen Fachgesellschaften und Patientenorganisationen in den Aufbau des Netzwerks und in die Erstellung der Leitlinien. Am Dr. von Haunerschen Kinderspital wird insbesondere eine moderne genetische Diagnostik wie Exom- und Genom-Sequenzierung ermöglicht. Häufig sind über die genetischen Untersuchungen hinaus auch zell- und molekularbiologische Validierungsuntersuchungen nötig, die ebenfalls im Sinne des ERN-RITA grenzübergreifend ermöglicht werden. Auch wenn die grenzüberschreitende Leistungsvergütung aktuell noch nicht geklärt ist, sollen mittelfristig die medizinischen Leistungen über eine harmonisierte europäische Vergütung abgebildet werden. Das explizite Ziel dieses ersten europäischen Gesundheitsprogramms besteht darin, allen Bürgerinnen und Bürgern der europäischen Union in grenzüberschreitender Weise eine vergleichbare Gesundheitsvor- und Fürsorge anzubieten.
Innovationsfond Krankenkassen
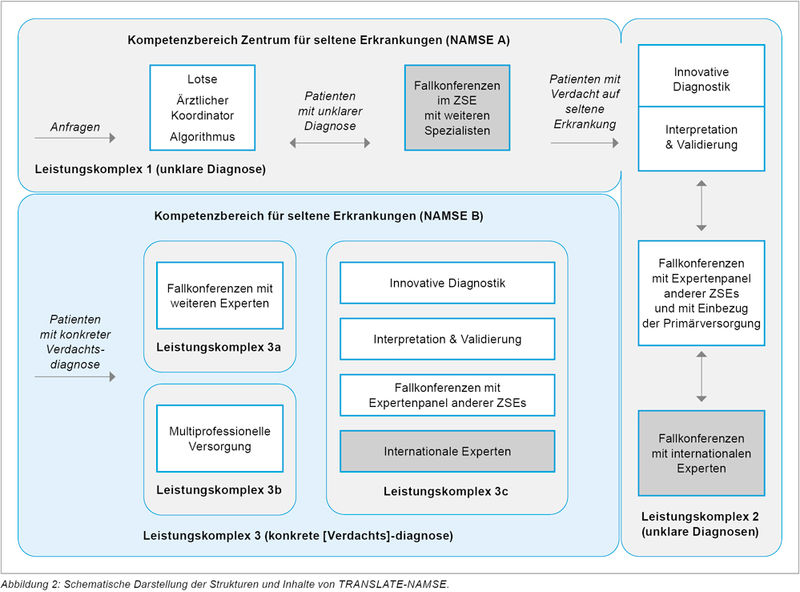
Auf nationaler Ebene hat der Innovationsfond der Krankenkassen neue Akzente für Patienten mit SE gesetzt. In Bayern ist das Klinikum der LMU über die Immunologie im Dr. von Haunerschen Kinderspital an diesem wichtigen Projekt beteiligt [18]. Translate-Namse ist ein Verbund von acht universitären ZSE mit den gesetzlichen Krankenkassen AOK Nordost und Barmer sowie der ACHSE [19]. Ziele des Projekts sind eine beschleunigte und präzise Diagnosestellung durch Zugang zu überregionaler, interdisziplinärer Kompetenz sowie zu innovativer genetischer Diagnostik, eine Verbesserung der sektorenübergreifenden Versorgung durch IT-gestützte Kommunikationsplattformen und ein strukturierter Übergang von Patienten aus der Kinder- und Jungendmedizin in die Erwachsenenmedizin zur Vermeidung von Versorgungsbrüchen [19]. Die dazu im Projekt etablierten Maßnahmen sind der Einsatz von Lotsen und ärztlichen Koordinatoren an den NAMSE-Typ-A-Zentren, ein strukturiertes Fallmanagement und standardisierte Behandlungspfade in sogenannten Leistungskomplexen an den NAMSE-Typ-B-Zentren, sowie interdisziplinäre und multizentrische Fallkonferenzen (Abbildung 2).
Der gezielten Durchführung innovativer genetischer Diagnostik kommt in Translate-Namse ein besonderer Stellenwert zu und im Laufe des Projektes ist es für die Bundesrepublik Deutschland erstmals gelungen, eine von den am Projekt beteiligten gesetzlichen Krankenkassen aus dem Innovationsfonds finanzierte Exomdiagnostik durchzuführen. Für die diagnostische Versorgung von Menschen mit SE bedeutet dies einen erheblichen Gewinn. Nicht nur kann die Zeit bis zur korrekten Diagnose deutlich verkürzt werden, auch die Diagnoserate wurde auf beeindruckende Weise verbessert. Eine umfassende quantitative und qualitative Auswertung der Translate-Namse-Daten soll im Herbst 2020 veröffentlicht werden.
Bayerische Allianz
Auf bayerischer Ebene wurde vor einigen Jahren die Bayerische Allianz für Seltene Erkrankungen (BASE) gegründet. BASE, ein Zusammenschluss der Zentren für SE der Universitätsklinika in München, Regensburg, Erlangen, Augsburg und Würzburg, wird aktuell von der Bayerischen Staatsregierung unterstützt, um auch im Flächenland Bayern neue Strukturen der Diagnostik und Therapie von Patienten mit SE zu fördern.
Fallbeispiele
Folgende Fallvignetten veranschaulichen anhand konkreter Beispiele die Arbeitsweise in den Projekten ERN-RITA und Translate-Namse und die daraus resultierende Verbesserung in der Versorgung von Menschen mit SE. Im Jahr 2017 beschrieben wir mit der CARMIL2-Defizienz eine neue Immundefekterkrankung, die neben den für einen kombinierten Immundefekt typischen infektiologischen Komplikationen insbesondere auch Epstein-Barr-Virus-assoziierten Weichteiltumoren (EBV+ smooth muscle tumors, EBV+ SMT) zeigte [20]. Zuvor waren EBV+ SMT insbesondere bei sekundärer Immundefizienz im Rahmen von HIV-Infektion und immunsuppressiver Behandlung nach Organtransplantation bekannt. Über ERN-RITA konnten wir im weiteren Verlauf an die 50 Menschen mit CARMIL2-Defizienz identifizieren, von denen 25 Prozent an bisher nicht behandelbaren und somit infausten EBV+ SMT erkrankt waren (Hauck et al., unveröffentlichte Daten). Mit Hilfe der via CPMS kommunizierenden Expertenpanels konnten wir die allogene hämatopoietische Stammzelltransplantation als kurativen Ansatz konsentieren und inzwischen einen 16-jährigen Jugendlichen mit CARMIL2-Defizienz erfolgreich behandeln (Hauck et al., unveröffentlichte Daten). Ein weiteres krankes Kind wird aktuell transplantiert und zwei Eingriffe befinden sich in der Vorbereitung.
Im Rahmen des Translate-Namse-Projekts wurde in einer interdisziplinären Fallkonferenz aus pädiatrischen Neurologen, Rheumatologen, Immunologen und Humangenetikern ein sechs Jahre altes Mädchen mit Zustand nach Hirnstamminfarkt und im weiteren Verlauf manifestierendem Erythema nodosum, Livedo racemosa und Raynaud-Phänomen besprochen. Aufgrund der Verdachtsdiagose einer SE aus dem Formenkreis der Vaskulitiden wurde eine Exom-Sequenzierung durchgeführt. Im Ergebnis sahen wir gekoppelt heterozygote Varianten im ADA2-Gen. Dieser genetische Befund konnte anschließend funktionell validiert werden und die Diagnose somit gesichert werden (Hauck et al., unveröffentlichte Daten). Bei der ADA2-Defizienz handelt es sich um eine autoinflammatorische Störung, die neben wiederkehrender Vaskultitis mit teilweise hämorrhagischen Schlaganfällen auch zu einem Immunglobulinmangel und zu einer Neutrozytopenie führen kann [21]. Da die Entzündungsreaktion bei ADA2-Defizienz insbesondere über den Tumornekrosefaktor-alpha (TNF-α) vermittelt wird, kann die Störung gut mit dem TNF-α-Antagonisten Etanercept außerhalb der Zulassung behandelt werden [22]. Nach dem Einholen der entsprechenden Behandlungseinwilligung und der Kostenübernahme ist die pädiatrische Patientin nun unter Etanercept beschwerdefrei und hat nach aktuellem Wissensstand ein deutlich gesenktes Risiko einen weiteren Schlaganfall zu erleiden.
Zusammenfassung
Zusammenfassend rücken SE als Krankheitsgruppe aufgrund ihrer großen Bedeutung für die Populationsgesundheit zunehmend in das Bewusstsein medizin-politischer Aktivitäten. Zahlreiche klinisch-translationale Projekte sind entstanden, neue Versorgungsstrukturen und -inhalte werden gestaltet. Bisher unveröffentlichte Daten zeigen einen hohen Mehrwert einer frühzeitigen modernen genetischen Diagnostik nicht nur für Kinder- und Jugendliche, sondern auch für Erwachsene mit SE. Daher gilt es nun, die Strukturen der ZSE nachhaltig zu sichern und eine leistungsorientierte und grenzüberschreitende Vergütung für die ZSE zu etablieren.
Das Literaturverzeichnis kann im Internet unter www.bayerisches-aerzteblatt.de (Aktuelles Heft) abgerufen werden.
Autoren
Privatdozent Dr. Dr. sci. nat. Fabian Hauck1, 2, 3
Antonia Pelshenke1, 2
Dr. Stella Bergemann1, 2
Julia Eilenberger1, 2
Dr. Maximilian Witzel1, 2
Professor Dr. Michael Albert1, 2
Professor Dr. Dr. sci. nat. Christoph Klein1, 2, 3
1 Kinderklinik und Kinderpoliklinik im Dr. von Haunerschen Kinderspital, Klinikum der Universität München, Ludwig-Maximilians-Universität München
2 Münchner Zentrum für Seltene Erkrankungen, Care-for-Rare Center am Dr. von Haunerschen Kinderspital, Klinikum der Universität München, Ludwig-Maximilians-Universität München
3 Deutsches Zentrum für Infektionsforschung, Standort Ludwig-Maximilians-Universität München.
Korrespondenzadresse:
Privatdozdent Dr. Dr. sci. nat. Fabian Hauck, Immundefektambulanz,
Dr. von Haunersches Kinderspital,
Lindwurmstraße 4, 80337 München,
Tel. 089 4400-53931, Fax 089 4400-53964,
E-Mail: fabian.hauck(at)med.uni-muenchen.de
Teilen:
Das könnte Sie auch interessieren: