02.12.2025
Titelthema
![]() Artikel als PDF
Artikel als PDF
Neu in der Pathologie
 Pathologie
Pathologie
Prädiktive und Prognostische Biomarker für gastrointestinale Tumoren
Die aktuellen diagnostischen und therapeutischen Entwicklungen haben die Versorgung von Patientinnen und Patienten mit gastrointestinalen Tumoren in den vergangenen Jahren erheblich verbessert. Die Pathologie spielt dabei eine zentrale Rolle: Molekulare Marker wie CLDN18.2 beim Magenkarzinom ermöglichen eine gezielte Antikörpertherapie mit Zolbetuximab, während die Bestimmung der Mikrosatelliteninstabilität (MSI) beim kolorektalen Karzinom einen prädiktiven Marker für den Einsatz einer Immuntherapie darstellt und eine frühzeitige Risikostratifizierung ermöglicht. Für frühe kolorektale Karzinome (pT1) bringt die neue S3-Leitlinie eine wichtige Änderung: Die Submukosainvasion allein gilt nicht mehr als Hochrisikofaktor für das nodale Metastasierungsrisiko, sofern Differenzierungsgrad, Lymphgefäßinvasion, Resektionsstatus und Tumor-Budding unauffällig sind. Interdisziplinäre Befundbewertung durch Pathologie, Gastroenterologie und Chirurgie ist hier entscheidend, um eine Über- oder Untertherapie zu vermeiden. Das molekulare Tumorboard bündelt diese interdisziplinären Expertisen, bewertet die komplexen Befunde gemeinsam und unterstützt so die präzise, patientenorientierte Therapieentscheidung.
Fall 1
Patient M., 62 Jahre, wird wegen unspezifischer Oberbauchbeschwerden gastroskopiert. Es zeigt sich ein ulzerierender Tumor an der kleinen Kurvatur des Magens. Die Biopsie bestätigt ein tubuläres Adenokarzinom vom intestinalen Typ nach Laurén. Die Immunhistochemie zeigt eine starke membranöse Claudin 18.2-Expression in 90 Prozent der Tumorzellen und ein negatives Färbeergebnis in der HER2-Immunhistochemie (Score 0). Aufgrund dieser Markerkonstellation wird eine Therapie mit Zolbetuximab in Kombination mit platin- und fluoropyrimidinhaltiger Chemotherapie empfohlen. Der Patient erhält zunächst eine systemische Therapie, um die Tumormasse zu reduzieren, und wird anschließend interdisziplinär für eine mögliche chirurgische Resektion evaluiert.
CLDN18.2 als neuer Biomarker beim Magenkarzinom – Grundlage für die Therapie mit Zolbetuximab
Das Magenkarzinom zählt weltweit zu den häufigsten und tödlichsten Tumorerkrankungen. Lange Zeit standen für die systemische Behandlung lediglich Chemotherapie [1] und, bei HER2-positiven Tumoren, Trastuzumab zur Verfügung. Mit Zolbetuximab ist nun erstmals ein Antikörper für eine weitere klar definierte Patientengruppe zugelassen: für Patienten mit fortgeschrittenem oder metastasiertem, HER2-negativem Adenokarzinom des Magens oder des gastroösophagealen Übergangs, deren Tumoren den Marker Claudin 18.2 (CLDN18.2) in ausreichendem Maß exprimieren. In der bislang größten untersuchten Fallserie zeigte sich eine CLDN18.2-Prävalenz von 38,4 Prozent bei Magen- und gastroösophagealen Adenokarzinomen [2].
Claudine sind Strukturelemente der sogenannten „tight junctions“, die die Barrierefunktion von Epithelzellen sichern. Die Isoform „Claudin 18.2“ kommt nahezu ausschließlich in der Magenschleimhaut vor. Im gesunden Gewebe ist sie fest in den Zell-Zell-Kontakten verankert und für den therapeutischen Antikörper schwer zugänglich. Im Zuge der malignen Transformation verliert das Gewebe diese enge Architektur; dadurch werden die Epitope von Claudin 18.2 auf der Zelloberfläche für therapeutische Antikörper erreichbar. Zolbetuximab bindet hochspezifisch an CLDN18.2 und führt über zwei immunologische Mechanismen zur Tumorzellzerstörung: zum einen vermittelt es eine antikörperabhängige zelluläre Zytotoxizität durch natürliche Killerzellen, zum anderen aktiviert es das Komplementsystem mit nachfolgender Zelllyse. In Kombination mit einer platin- und fluoropyrimidinhaltigen Chemotherapie konnte in großen Phase-3-Studien eine signifikante Verlängerung des progressionsfreien und des Gesamtüberlebens nachgewiesen werden. In der SPOTLIGHT-Studie lag der Anteil von überlebenden Patienten nach 36 Monaten bei 20,9 Prozent im Zolbetuximab- und bei 13,7 Prozent im Placebo-Arm, in der GLOW-Studie bei 18,3 Prozent vs. 7,9 Prozent [4, 5].
Für die Indikationsstellung ist eine zuverlässige Immunhistochemie (IHC) unverzichtbar. Mit ihr wird geprüft, ob ein Tumor CLDN18.2 in relevantem Umfang exprimiert. Das Färbeergebnis wird nach einem einheitlichen Schema beurteilt, das auch den Zulassungsstudien zugrunde lag: Als CLDN18.2-positiv gelten Tumoren dann, wenn mindestens 75 Prozent der Tumorzellen eine membranöse Färbung mit mindestens mäßiger bis starker Intensität aufweisen (Abbildung 1). Rein zytoplasmatische oder schwache Signale gelten nicht als spezifisch. Gerade Fälle mit heterogener oder grenzwertiger Expression erfordern eine besonders sorgfältige Beurteilung, da falsch-negative Ergebnisse den Zugang zu einer wirksamen Therapie versperren können [6].

Abbildung 1: Claudin 18.2 Expression in normaler Magenschleimhaut (A) und in Adenokarzinomen des Magens (B bis E). Panel B zeigt einen komplett negativen Fall. Magenkarzinome mit geringer (C), mäßiger (D) und starker (E) Färbeintensität. Eine Claudin 18.2-Positivität wird als eine zumindest mäßige bis starke membranständige Expression in ≥ 75 % der Tumorzellen definiert.
Die Bedeutung einer hohen Testqualität wurde jüngst in einem Ringversuch in Deutschland, Österreich und der Schweiz unterstrichen. Dort zeigte sich, dass die Mehrheit der beteiligten Labore konsistente Ergebnisse erzielte, es aber bei einzelnen Färbeprotokollen zu Problemen mit der Sensitivität kam. Insgesamt verdeutlicht die Studie, dass eine standardisierte Vorgehensweise und die regelmäßige Qualitätssicherung entscheidend sind, um eine sichere Patientenselektion zu gewährleisten [7].
Damit etabliert sich CLDN18.2 als neuer diagnostischer Standard in der Pathologie und zugleich als Schlüsselfaktor für die Therapieentscheidung. Für die klinische Praxis bedeutet dies: Nur Patienten mit eindeutig positivem Test profitieren von Zolbetuximab. Die enge Zusammenarbeit zwischen Pathologie und behandelnden Ärztinnen und Ärzten ist daher unverzichtbar, um die Indikationsstellung korrekt und rechtzeitig zu stellen. Mit der Etablierung dieser Testung eröffnet sich für viele Betroffene mit fortgeschrittenem Magenkarzinom erstmals eine zusätzliche therapeutische Option.
Fall 2
Patientin S., 58 Jahre, stellt sich mit Blutbeimengungen im Stuhl vor. Die Koloskopie zeigt ein tumoröses Geschehen im Sigma, die Biopsie bestätigt ein Adenokarzinom. Immunhistochemie (Ausfall von MLH1 und PMS2) und PCR-Untersuchung zeigen eine hochgradige Mikrosatelliteninstabilität (MSI-H) ohne BRAF-Mutation (Tabelle 1) und ohne MLH1-Promotor-Methylierung, was auf eine mögliche Assoziation des Karzinoms mit einem Lynch-Syndrom hinweist (Autosomal dominant vererbte Prädisposition, die durch Mutationen in DNA-Mismatch-Repair-Genen verursacht wird und beim Dickdarmkarzinom zu einem erhöhten Risiko für frühzeitiges Auftreten und charakteristische Mikrosatelliteninstabilität [MSI] führt). Nach interdisziplinärer Beratung erfolgt eine rechtsseitige Hemikolektomie mit Lymphadenektomie. Aufgrund des MSI-H-Status wird die Patientin nach der Operation für eine Immuntherapie evaluiert und erhält zudem eine genetische Beratung zur Abklärung einer erblichen Prädisposition.
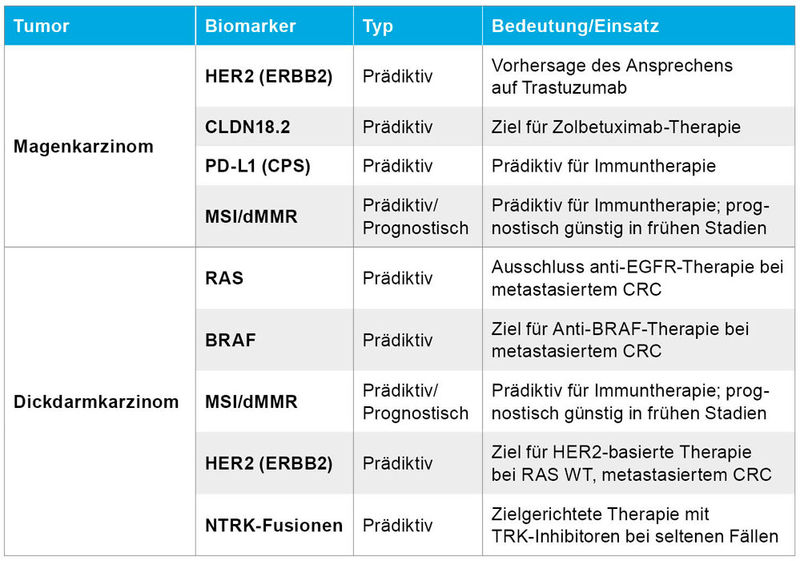
Tabelle 1: Prädiktive- und prognostische Biomarker bei gastrointestinalen Tumoren [14].
Mikrosatelliteninstabilität (MSI) beim Dickdarmkarzinom – neue Empfehlung der S3-Leitlinie
Die im September 2025 veröffentlichte Aktualisierung der S3-Leitlinie für das kolorektale Karzinom bringt einige Neuerungen für die klinische Praxis. Erstmals wird empfohlen, bei allen Patienten mit neu diagnostiziertem Dickdarmkarzinom den MSI-Status zu bestimmen [8].
Tumoren mit hochgradiger Mikrosatelliteninstabilität (MSI-H) unterscheiden sich in ihrem biologischen Verhalten deutlich von mikrosatellitenstabilen (MSS) Karzinomen. In den UICC-Stadien I bis III haben MSI-H-Karzinome eine deutlich günstigere Prognose: Sie entwickeln seltener Lymphknoten- und Fernmetastasen. So beträgt die Fernmetastasierungsrate bei MSI-H-Tumoren nur rund 8 Prozent, während sie bei MSS-Tumoren etwa 27 Prozent beträgt. In fortgeschrittenen Stadien (UICC IV) hingegen ist eine MSI-H seltener, tritt nur in 4 bis 5 Prozent der Fälle auf und ist in dieser Gruppe mit einer schlechteren Prognose verknüpft, was unter anderem auf die häufigere Assoziation mit BRAF-Mutationen zurückzuführen ist [9].
Unabhängig vom Tumorstadium liefert der MSI-Status entscheidende prädiktive Informationen: MSI-H-Tumoren reagieren schlechter auf eine klassische adjuvante Chemotherapie, sprechen aber besonders gut auf immunonkologische Therapien an. Diese Therapieansätze nutzen die hohe Tumor-Mutationslast von MSI-H-Tumoren, die zu einer verstärkten Bildung von Neoantigenen und einer ausgeprägten T-Zell-Antwort führt. Monoklonale Antikörper gegen PD-1, PD-L1 oder CTLA-4 können die tumorinduzierte Immun-Resistenz durchbrechen und eine gezielte Zerstörung von Tumorzellen durch zytotoxische Mechanismen induzieren [10].
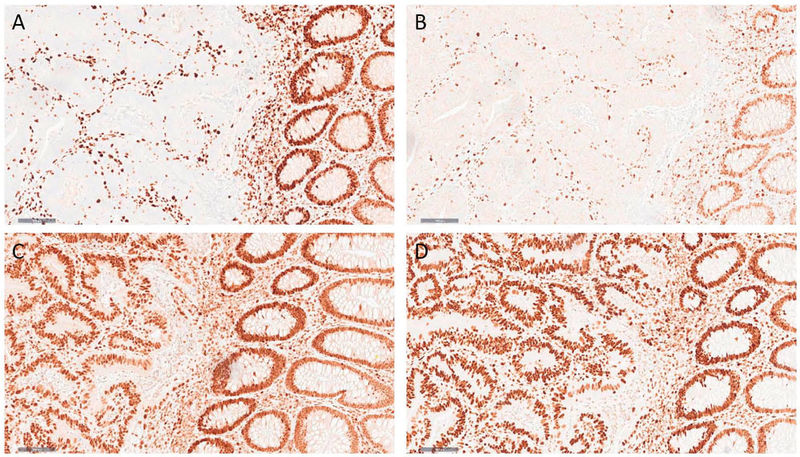
Abbildung 2: Dickdarmkarzinom mit defizientem Mismatch-Reparatur-Status (dMMR). In der Immunhistochemie für MLH1 (A) und PMS2 (B) zeigt sich ein Verlust der nukleären Proteinexpression bei regelhaftem Ausfall der endogenen Kontrollen (Lymphozyten und normale Kolonschleimhaut) bei denen sich eine erhaltene Proteinexpression zeigt. Die Färbungen zum Nachweis von MSH2 (C) und MSH6 (D) zeigen eine erhaltene Proteinexpression in den Tumorzellkernen.
Für die Diagnostik in der Pathologie steht die Immunhistochemie im Mittelpunkt. Dabei werden die Proteine des Mismatch-Reparatur-Systems (MLH1, MSH2, MSH6, PMS2) direkt im Tumorgewebe angefärbt (Abbildung 2). Fehlt die nukleäre Expression eines oder mehrerer Proteine, spricht dies für eine MSI-H bzw. einen Mismatch-Reparaturdefekt (dMMR). Diese Methode ist etabliert, schnell durchführbar und als Routineuntersuchung in der Pathologie weit verbreitet. Ergänzend kann die molekulare Analyse mittels PCR und Fragmentlängen-Analyse erfolgen, um Längenveränderungen in den Mikrosatelliten zu bestätigen.
Bei unklaren oder nicht plausiblen immunhistochemischen Befunden empfiehlt die Leitlinie die molekulare Bestätigung des MSI-Status, zum Beispiel durch PCR-basierte Analyse von definierten Mikrosatellitenmarkern. Darüber hinaus dient die molekulare Testung auch der Abklärung eines möglichen Lynch-Syndroms (Abbildung 3). So kann die Kombination aus BRAF-V600E-Mutation (Abbildung 3 B) und MLH1-Promotor-Methylierung (Abbildung 3 C) helfen, sporadische MSI-H-Tumoren von Lynch-assoziierten Tumoren zu unterscheiden: Eine BRAF-Mutation oder eine MLH1-Methylierung spricht in der Regel gegen ein Lynch-Syndrom. Liegt keine BRAF-Mutation oder Promotor-Methylierung vor, sollte eine humangenetische Beratung erfolgen, um das Vorliegen einer erblichen Prädisposition abzuklären [11].
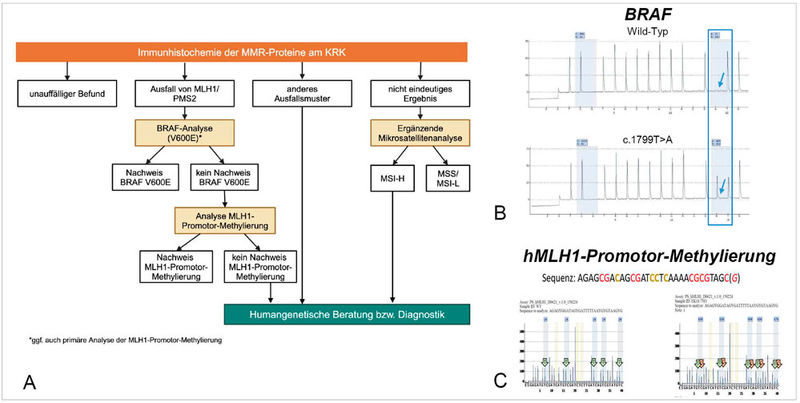
Abbildung 3: Molekularer Testalgorithmus zur Abklärung eines möglichen Lynch-Syndroms (A). Der Nachweis einer BRAF-Mutation, hier mittels Pyrosequenzierung (B) und/oder einer Hypermethylierung der MLH1-Promotor-Region (C) sprechen für eine sporadische Genese des Dickdarmkarzinoms. Modifiziert nach [8].
Die neue S3-Leitlinie schlägt hierfür einen klar strukturierten Algorithmus vor, der Pathologinnen und Pathologen eine standardisierte Vorgehensweise zur Interpretation der immunhistochemischen und molekularen Befunde an die Hand gibt (Abbildung 3 A). Dies erleichtert die frühzeitige Identifikation von Hochrisikopatienten und die zielgerichtete Weiterbehandlung [8].
Die neue Leitlinienempfehlung, MSI bereits bei Erstdiagnose routinemäßig zu testen, ermöglicht eine frühzeitige Risikostratifizierung und eine gezielte Therapieauswahl. Dies stellt einen bedeutenden Fortschritt für die Versorgung von Patienten mit Dickdarmkarzinom dar.
Aktuelles Polypenmanagement im Kolon: Neuerungen der S3-Leitlinie
Auch das Polypenmanagement im Kolon wurde mit der neuen Fassung der S3-Leitlinie weiterentwickelt (Tabelle 2). Für konventionelle Adenome gelten abgestufte Surveillance-Intervalle: Nach Abtragung von ein bis zwei kleinen, unauffälligen Adenomen (< 1 cm, tubulär, ohne hochgradige Dysplasie) genügt eine Kontrollkoloskopie nach sieben bis zehn Jahren. Bei höherem Risiko (drei bis vier Adenome, Größe < 1 cm, ohne villöse Architektur oder hochgradige intraepitheliale Neoplasie) wird ein Intervall von drei bis fünf Jahren empfohlen. Bei großen Adenomen (≥ 1 cm) oder Nachweis einer villösen Architektur oder hochgradiger intraepithelialer Neoplasie sollte bereits nach drei Jahren eine Kontrollendoskopie erfolgen. Große oder sessile Läsionen (≥ 20 mm) sollen möglichst en-bloc oder im Piecemeal-Verfahren vollständig entfernt werden; hier ist eine frühzeitige Kontrollkoloskopie nach sechs Monaten sinnvoll, um Residuen auszuschließen [12].
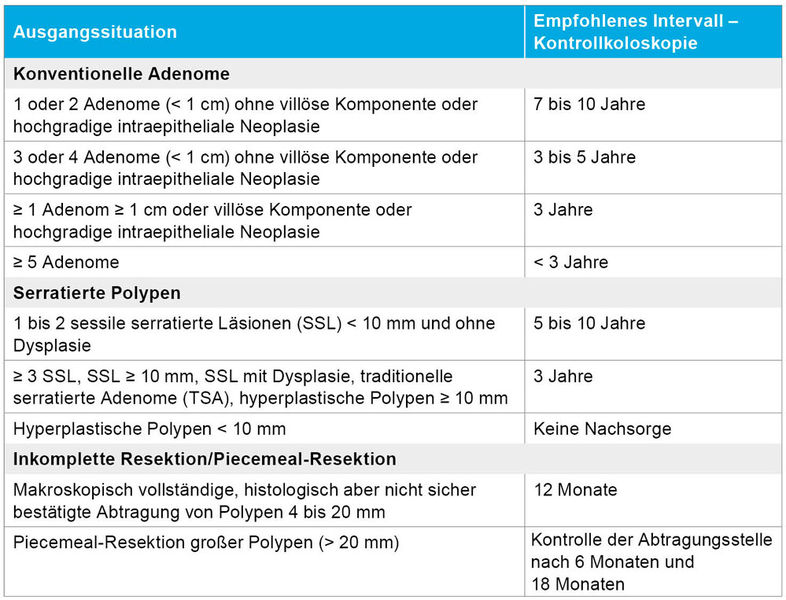
Tabelle 2: Empfohlene Surveillance-Intervalle für Dickdarmpolypen nach der neuen S3-Leitlinie für das Dickdarmkarzinom.
Mehr in den Fokus rücken auch wieder die serratierten Polypen (sessile serratierte Läsionen - SSL; traditionelle serratierte Adenome – TSA). Während kleine hyperplastische Polypen im Rektosigmoid meist harmlos sind und keine Surveillance erfordern, werden SSL ≥ 10 mm oder SSL mit Dysplasie sowie alle TSA als fortgeschrittene Läsionen eingestuft. Nach kompletter Entfernung ist hier eine Kontrollkoloskopie nach drei Jahren vorgesehen, bei multiplen oder unvollständig resezierten Läsionen gegebenenfalls früher [8].
Fall 3
Patient K., 65 Jahre, wird aufgrund eines positiven Hämoccult-Tests koloskopiert. In der rektosigmoidalen Region wird ein 1,2 cm großer Polyp entdeckt und endoskopisch abgetragen. Die histologische Aufarbeitung zeigt ein wenig differenziertes Adenokarzinom mit Lymphgefäßinvasion (L1) und Tumor-Budding Grad 3. Die Submukosainvasion beträgt 1.200 µm. Der Resektionsrand ist tumorfrei (R0). Aufgrund der hohen Risiko-Konstellation für nodale Metastasen wird nach interdisziplinärer Diskussion eine segmentale Kolektomie mit systematischer Lymphknotenentfernung durchgeführt. In zwei von 25 entfernten Lymphknoten wurden Metastasen nachgewiesen, was die Indikation zur Kolektomie bestätigte.
Frühe kolorektale Karzinome (pT1) und Risikostratifizierung
Im Dickdarm wird ein invasives Karzinom erst diagnostiziert, wenn Tumorzellformationen in die Submukosa einwachsen. Dies unterscheidet sich von anderen Abschnitten des Gastrointestinaltrakts, in denen bereits ein Durchbruch durch die Basalmembran und eine Infiltration der Lamina propria als Karzinom gelten. Die lokale Tumorinvasion ist eng mit dem Risiko einer loko-regionären lymphogenen Metastasierung verknüpft. Die aktuelle Fassung der S3-Leitlinie zum kolorektalen Karzinom empfiehlt, pT1-Karzinome zusätzlich nach ihrem nodalen Metastasierungsrisiko in Low-risk- und High-risk-Fälle einzuteilen. Neu ist dabei, dass die Tiefe der Submukosainvasion allein – selbst wenn sie > 1.000 µm beträgt – nicht mehr als Hochrisikofaktor gilt, sofern alle anderen Parameter unauffällig sind. Low-risk-Karzinome umfassen vollständig in sano resezierte (R0), gut- bis mäßig-differenzierte Adenokarzinome (G1/G2 bzw. low grade nach WHO), ohne Lymphgefäßinvasion (L0) und ohne wesentliches Tumor-Budding (Budding = Einzelne Tumorzellen oder kleine Zellgruppen am Tumorrand, die auf eine aggressive Tumorbiologie und höheres Metastasierungsrisiko hinweisen). In dieser Konstellation liegt das Risiko einer lymphogenen Metastasierung nach vollständiger Tumorentfernung deutlich unter 6 Prozent [12].
High-risk-Fälle zeichnen sich hingegen durch folgendes histopathologisches Risikoprofil aus: schlecht differenzierte oder undifferenzierte Karzinome (G3/G4 bzw. high grade nach WHO), Tumoren mit Lymphgefäßinvasion (L1) oder unvollständig entfernte Tumoren (R1/R2). Auch ein Tumor-Budding Grad 2 bis 3 (> 4 Budds pro Gesichtsfeld) wird als relevanter Risikofaktor gewertet. In dieser Gruppe steigt das Risiko für nodale Metastasen auf etwa 20 Prozent [12].
Tumor-Budding (Der Begriff „Budding“ stammt aus dem Englischen und bedeutet „Knospe bilden“, da die Zellen wie kleine Knospen vom Tumorrand „abknospen“) beschreibt kleine Zellcluster von maximal fünf entdifferenzierten oder isolierten Tumorzellen an der Invasionsfront (Abbildung 4). Die Quantifizierung erfolgt mikroskopisch bei 200-facher Vergrößerung: Grad 1 umfasst 0 bis 4 Budds, Grad 2 umfasst 5 bis 9 und Grad 3 mehr als 9 Budds pro Sichtfeld. Zahlreiche Studien bestätigen, dass ein höherer Tumor-Budding-Grad mit einem signifikant erhöhten Risiko für Lymphknotenmetastasen einhergeht [13].
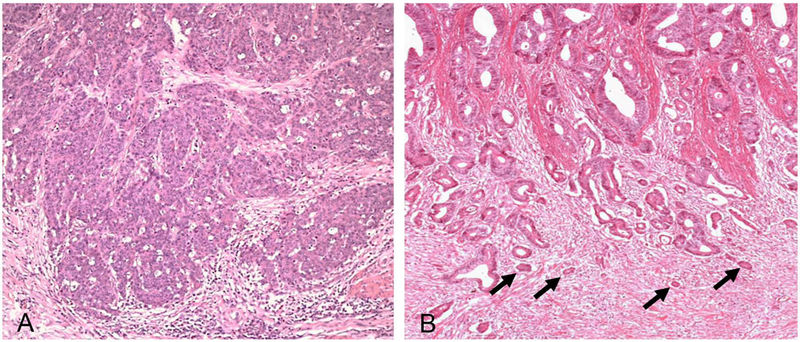
Abbildung 4: Tumor-Budding beim Dickdarmkarzinom. Panel A stellt den Befund eines gering ausgeprägten Tumor-Buddings dar, wohingegen das Panel B einen Fall mit hohem Tumor-Budding als Surrogat für eine starke Aktivierung des Wnt/β-catenin-Signalwegs zeigt. Die Pfeile markieren die kleinen Tumorzell-Knospen (sog. „Budds“).
Die differenzierte Risikobewertung hat direkte klinische Konsequenzen. Low-risk-pT1-Karzinome können häufig durch eine vollständige endoskopische Resektion mit nachfolgender Nachsorge behandelt werden. High-risk-Fälle erfordern eine onkologische Kolektomie mit systematischer Lymphknotenentfernung, um die onkologische Sicherheit zu gewährleisten. Die Leitlinie verdeutlicht damit, dass die Entscheidung über das weitere Vorgehen nicht mehr allein auf der Tiefe der Submukosainvasion beruhen sollte. Vielmehr werden mehrere Risikofaktoren herangezogen, um Patienten individuell zu stratifizieren. Die sorgfältige histopathologische Begutachtung – insbesondere Tumorgraduierung, Lymphgefäßinvasion, Residualstatus und Tumor-Budding – ist entscheidend [8]. Dies erfordert eine enge interdisziplinäre Zusammenarbeit zwischen Pathologie, Gastroenterologie und Chirurgie, um die optimale Therapieentscheidung zu treffen und Über- oder Untertherapie zu vermeiden.
Das molekulare Tumorboard bündelt die Expertise dieser Fachrichtungen, bewertet gemeinsam die komplexen molekularen Befunde und unterstützt so die präzise, patientenorientierte Auswahl individueller Therapieoptionen. Angesichts der zunehmenden Bedeutung molekularer Diagnostik und neuer Therapiestrategien wird die Rolle des molekularen Tumorboards in Zukunft voraussichtlich weiter an Gewicht gewinnen und eine noch zentralere Funktion in der Therapieplanung gastrointestinaler Tumoren einnehmen.
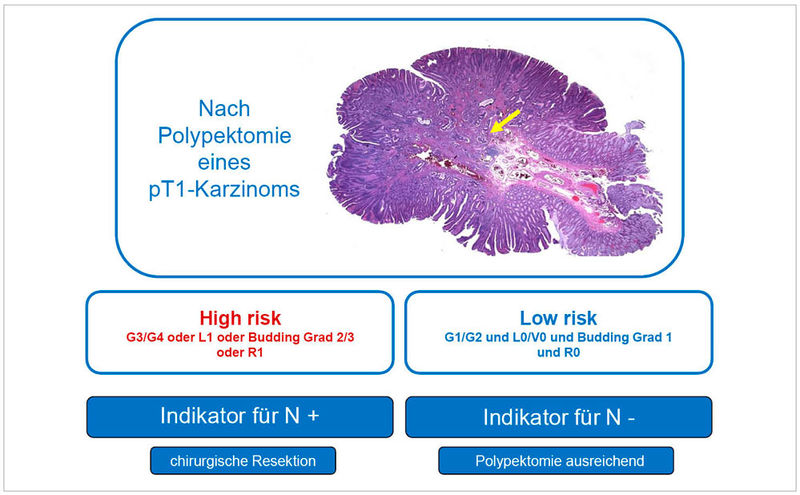
Abbildung 5: Schema für die Risikostratifizierung für pT1 kolorektale Karzinome. Der gelbe Pfeil markiert ein invasives Karzinom auf dem Boden eines großen gestielten tubulären Adenoms mit teils leichtgradiger und teils schwerer Epitheldysplasie. Sowohl das Karzinom als auch das Adenom sind in sano reseziert.
Das Wichtigste in Kürze
CLDN18.2 beim Magenkarzinom und MSI beim Dickdarmkarzinom sind entscheidende Biomarker für Diagnostik und Therapie. In der neuen S3-Leitlinie wird bei frühen kolorektalen Karzinomen die Tiefe der Submukosainvasion nicht mehr als Risikokriterium aufgeführt. Stattdessen fließen die Tumorgraduierung, die Lymphgefäßinvasion, der Residualstatus und das Tumor-Budding in die Risikoeinschätzung ein. Eine sorgfältige histopathologische Analyse und standardisierte Testverfahren sind entscheidend, um Patientinnen und Patienten gezielt zu therapieren und Über- oder Untertherapie zu vermeiden.
Der Autor erklärt, dass er für Agilent, Astellas, BMS, Falk, MSD, Pierre Fabre, Pfizer und Roche Beratungstätigkeiten ausgeübt und Honorare für Vorträge erhalten hat.
Das Literaturverzeichnis kann im Internet unter www.bayerisches-aerzteblatt.de (Aktuelles Heft) abgerufen werden.
Autor:

Universitätsprofessor Dr. Dr. Jens H. L. Neumann
Direktor des Universitätsinstituts für Pathologie, Paracelsus Medizinische Privatuniversität, Klinikum Nürnberg,
Prof.-Ernst-Nathan-Str. 1, 90419 Nürnberg, E-Mail: jens.neumann@klinikum-nuernberg.de
Teilen:
Das könnte Sie auch interessieren: